DNA Photophysics
Students in the Kohler Group pioneered the study of the excited states of nucleic acids responsible for the UV damage of DNA using ultrafast laser spectroscopy. Experiments performed over two decades and the computational studies inspired by this work have greatly advanced fundamental understanding of nonradiative decay pathways in DNA.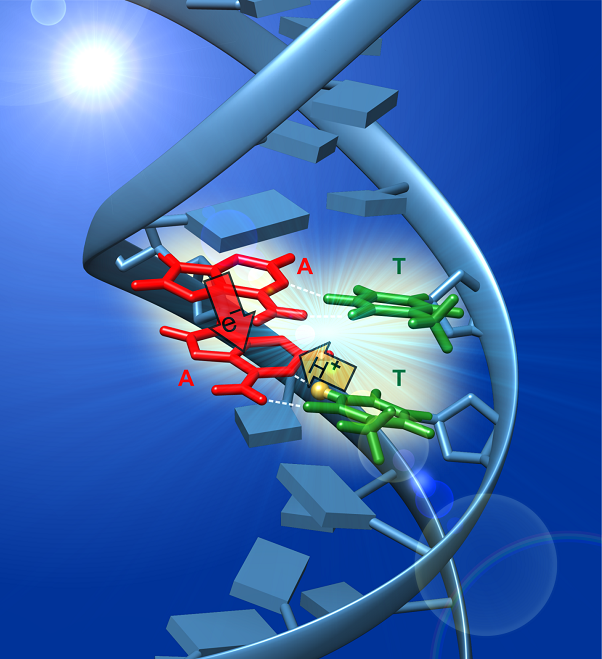
We collaborate actively with theorists, who carry out increasingly sophisticated quantum mechanical (QM) calculations on ever-larger DNA compounds. The combination of a tailorable multichromophoric molecule that is highly amenable to study by time-resolved spectroscopy and high-level quantum chemistry has put the field of DNA photophysics at the crossroads of frontier topics in chemical dynamics such as photoinduced proton-coupled electron transfer (PCET), nonadiabatic dynamics, and charge and exciton migration in supramolecular systems. Understanding the ultrafast dynamics of excited states in DNA and in hybrid DNA assemblies could lead to new strategies for directing energy or charge in multichromophoric systems made from organic building blocks, while minimizing undesired photochemical pathways that compromise photostability.
Following a bottom-up approach, we reported the first accurate measurements of excited state lifetimes (i.e., the fluorescence lifetimes) of single DNA and RNA nucleosides in aqueous solution [1]. These experiments revealed that all of the DNA and RNA bases have femtosecond lifetimes, which are thought to minimize the probability of UV photodamage to the genome, suggesting that the nucleobases are the legacy of photochemical selection that occurred before the emergence of life [2].
Unexpectedly, excited states of single- and double-stranded DNA oligonucleotides unexpectedly decay orders of magnitude more slowly than those of single bases [3]. The long-lived excited states are self-trapped excitons or excimers (exciplexes). The strong charge transfer character of these states is revealed by vibrational marker bands characteristic of nucleobase radical ions by femtosecond time-resolved IR (TRIR) spectroscopy [4].
Using TRIR spectroscopy, we have shown that oppositely charged base pair radical ions form in various double-stranded DNA molecules [5]. In several of the duplexes studied, an electron hops between bases on the same strand, attracting a proton to it from the opposite strand. This photoinduced PCET pathway results in the formation of a rare tautomeric base pair or distonic (“distant”) radical ion in which spin and charge reside on opposite DNA strands. These experiments are the first to observe the photoinduced transfer of a proton between the strands of the double helix.
Current work is focused on understanding how initially delocalized exciton states in DNA decay to charge-transfer excited states. Although light-harvesting proteins capture photon energy via delocalized excitons, the PCET events that take place subsequently involve thermally relaxed states that are located on molecular chromophores. We are exploring how ultrafast photoinduced PCET in DNA is influenced by the delocalized excitons present initially.
References
[1] Pecourt, J.; Peon, J.; Kohler, B. Ultrafast Internal Conversion of Electronically Excited RNA and DNA Nucleosides in Water. J. Am. Chem. Soc. 2000, 122, 9348-9349.
[2] Beckstead, A. A.; Zhang, Y.; de Vries, M. S.; Kohler, B. Life in the Light: Nucleic Acid Photoproperties as a Legacy of Chemical Evolution. Phys. Chem. Chem. Phys. 2016, 18, 24228-24238.
[3] Crespo-Hernández, C. E.; Cohen, B.; Kohler, B. Base Stacking Controls Excited-State Dynamics in A·T DNA. Nature 2005, 436, 1141-1144.
[4] Zhang, Y.; Dood, J.; Beckstead, A. A.; Li, X.-B.; Nguyen, K. V.; Burrows, C. J.; Improta, R.; Kohler, B. Efficient UV-Induced Charge Separation and Recombination in an 8-Oxoguanine-Containing Dinucleotide. Proc. Natl. Acad. Sci. U.S.A. 2014, 111, 11612-11617.
[5] Zhang, Y.; de La Harpe, K.; Beckstead, A. A.; Improta, R.; Kohler, B. UV-Induced Proton Transfer between DNA Strands. J. Am. Chem. Soc. 2015, 137, 7059-7062.
DNA-metal nanostructures
In this project, we exploit growing understanding of excited state dynamics in bare DNA to understand the photophysics of DNA coordinated to metal ions and metal nanoclusters. These structures are of interest for photonics applications and sensing. Metal coordination extends the noncovalent interactions of base stacking and hydrogen bonding found in DNA to produce photoactive, soft matter materials with fascinating structural and electronic properties.
Metal ions mediate new base pairing motifs, create supramolecular structures, and can even impart ‘metallic’ properties to DNA. Using the femtosecond transient absorption and time-resolved emission techniques, we are studying excited state dynamics in coordination polymers that form by self-assembly when silver(I) ions are combined with nucleobases such as adenine or 2-aminopurine. Atomic force microscopy (AFM) measurements indicate that the coordination polymers form nanofibrils several tens of nm in diameter that are many microns in length.
Chemically reducing silver ions bound to a DNA strand generates silver nanoclusters (AgNCs) containing no more than a few tens of atoms. These nanoclusters have remarkable optical properties such as tunable visible fluorescence and circular dichroism, but a model explaining their photophysical properties is lacking. We seek insights into the electronic interactions between AgNCs and their DNA hosts using time-resolved vibrational spectroscopy and other techniques.
DNA-metal structures are valuable model systems for fundamental studies of excited-stated dynamics at hybrid interfaces between organic and inorganic building blocks. Such interfaces are attractive for sensing and photocatalysis, and the insights obtained in this study could lead to new architectures for controlling the separation and recombination of photogenerated carriers and redox equivalents.