




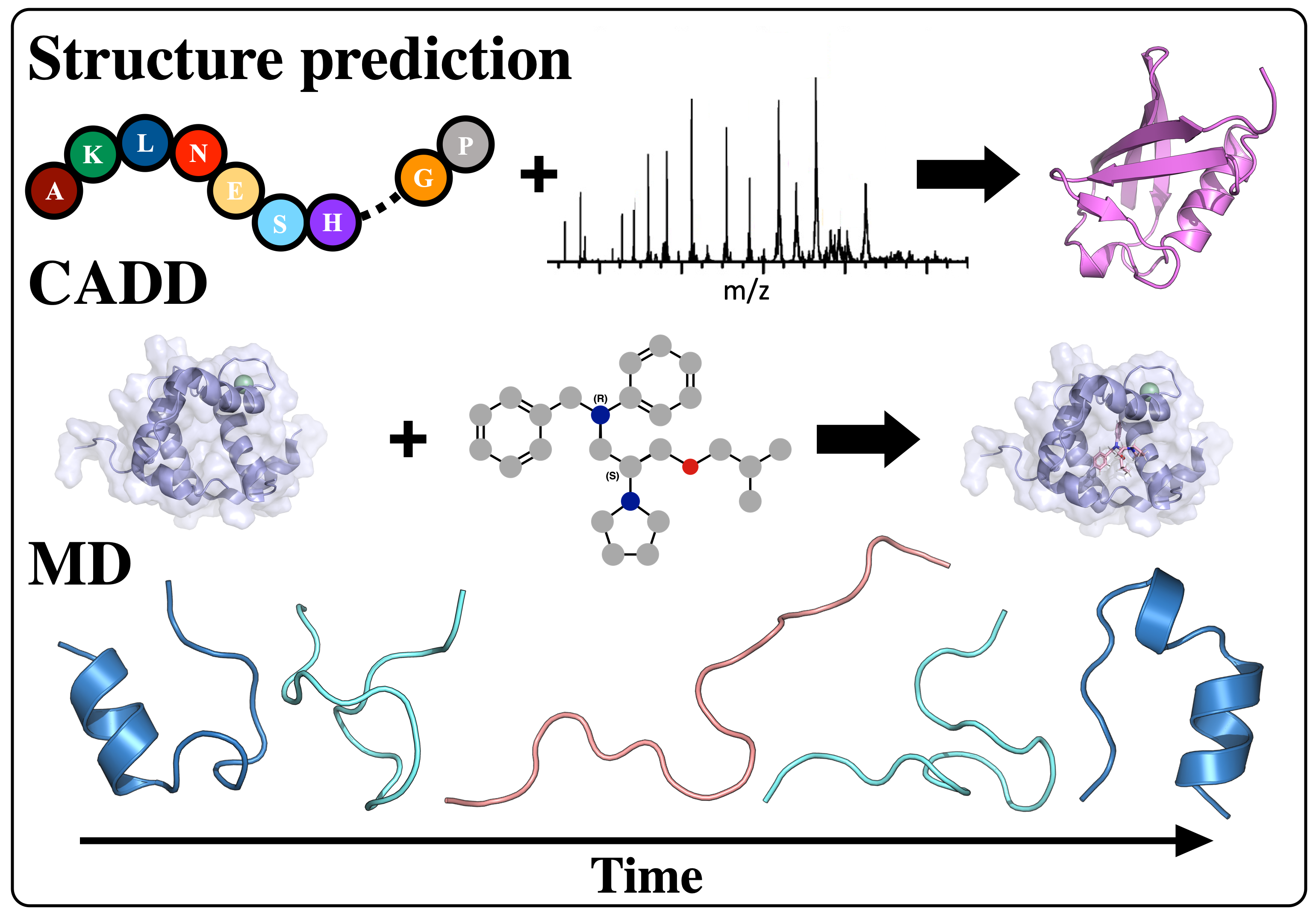
Computational Protein Structure Prediction from Sparse Experimental Data
Protein structure prediction is a highly successful computational method, offering a promising solution in cases where experimental techniques fail to determine protein structure. By combining sparse experimental data with computational modeling techniques, our lab has been able to make a notable contribution to this field. De novo structure prediction has been most successful when combined with sparse experimental data, forming a symbiosis where the combination of methods is more powerful than the sum of the individuals. The Lindert Lab is an active member of the Rosetta Commons Community, and we work to develop new algorithms for protein structure prediction which incorporate sparse experimental data from a variety of sources such as mass spectrometry, cryo-EM, and NMR. In recent years, artificial intelligence has made tremendous strides in protein structure prediction, and the lab is interested in incorporating machine learning/deep learning methods into all aspects of our current structure prediction projects.
Protein Structure Prediction Reviews:
Biehn, S.E.; Lindert, S. (2021), Protein Structure Prediction with Mass Spectrometry Data. Annu Rev Phys Chem. 73, 1-19.
Seffernick, J.T.; Lindert, S. (2020), Hybrid methods for combined experimental and computational determination of protein structure. J Chem Phys 153 (24), 240901.
Mass Spectrometry
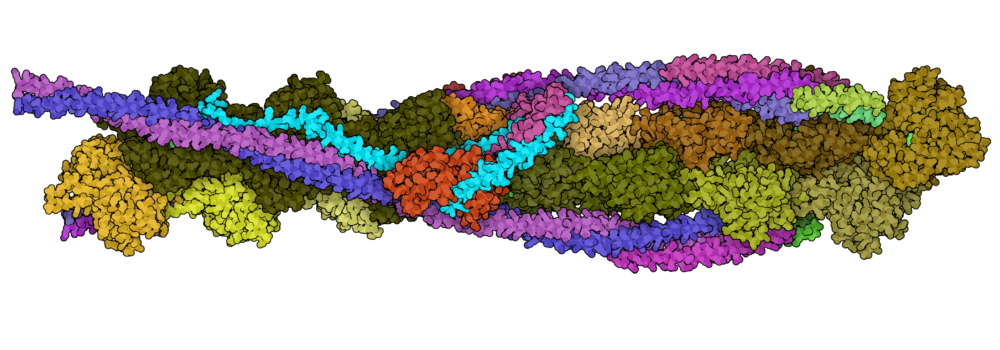
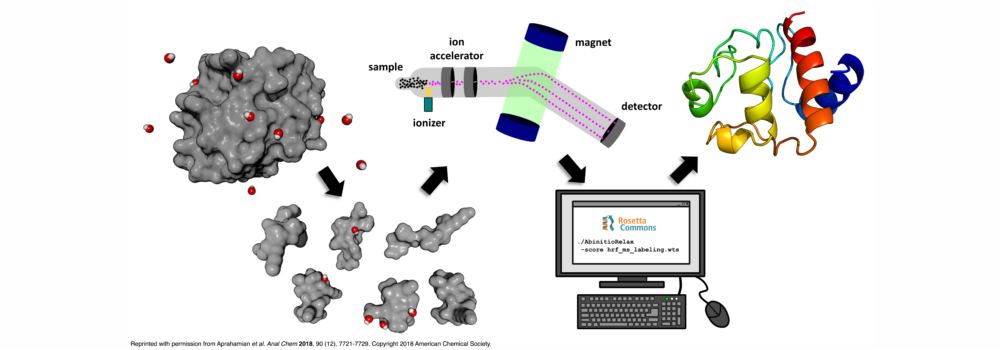
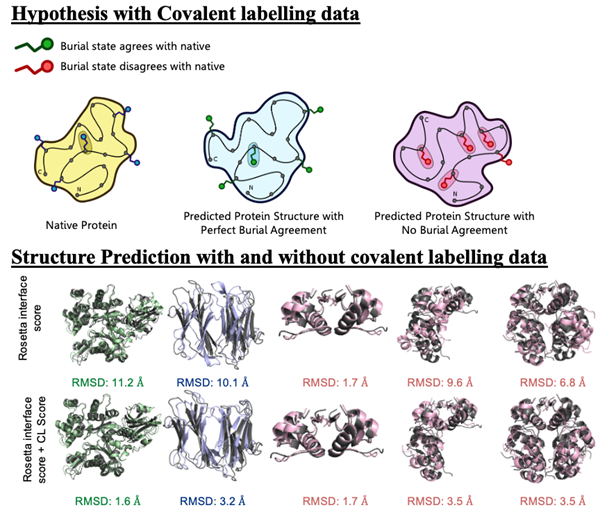
Mass spectrometry is an analytical technique that is used to measure the mass-to-charge ratio of ions. In very successful ongoing collaborations with Dr. Vicki Wysocki here at Ohio State University and other mass spetrometrists around the country, we have demonstrated the benefit of including sparse structural information from various mass spectrometry techniques. We have improved structure prediction algorithms by incorporating experimental data from covalent labeling (informing on residue solvent exposure), ion mobility (which informs on the shape and size of proteins), and surface-induced dissociation (which provides an insight to the sub-architecture of protein complexes).
Recent Related Publications:
Drake, Z.C.; Seffernick, J.T.; Lindert, S. (2022), Protein complex prediction using Rosetta, AlphaFold, and mass spectrometry covalent labeling. Nat Commun 13, 7846.
Turzo, S.B.A.; Seffernick, J.T.; Rolland, A.D.; Donor, M.T.; Heinze, S.; Prell, J.S.; Wysocki, V.H.; Lindert, S. (2022), Protein shape sampled by ion mobility mass spectrometry consistently improves protein structure prediction. Nat Commun 13 (1), 4377.
Biehn, S.E.; Lindert, S. (2021), Accurate Protein Structure Prediction with Hydroxyl Radical Protein Footprinting Data. Nat Commun 12, 341.
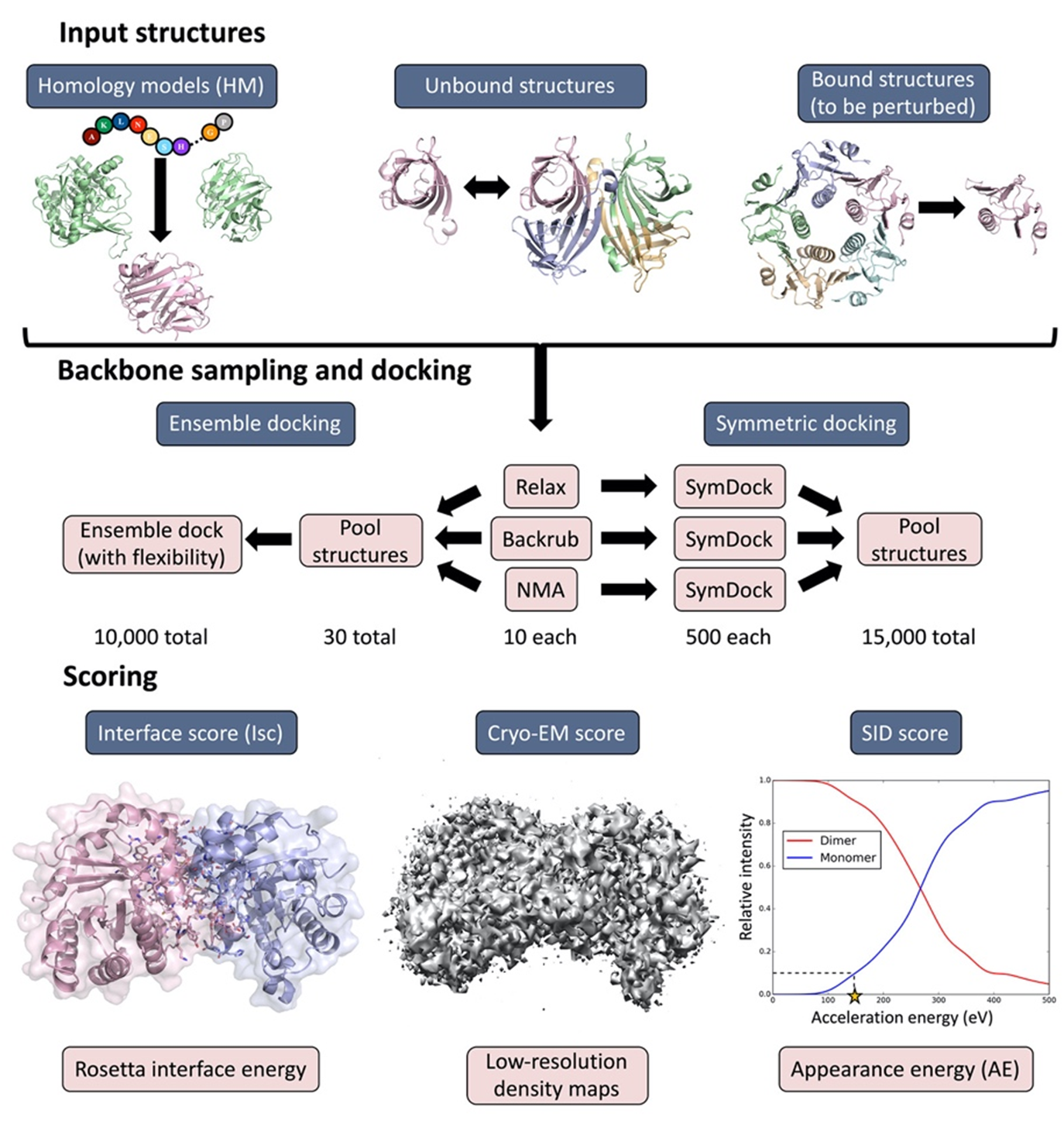
Seffernick, J.T.; Harvey, S.R.; Wysocki, V.H.; Lindert, S. (2019), Predicting Protein Complex Structure from Surface-Induced Dissociation Mass Spectrometry Data. ACS Cent Sci 5 (8), 1330-1341.
Cryogenic Electron Microscopy (cryo-EM)
Cryo-EM has become a primary method for protein structure elucidation, frequently yielding density maps with near-atomic or medium resolution. If protein structures cannot be determined unambiguously from the density maps, computational structure refinement tools are needed to generate protein structural models. We have demonstrated that using Rosetta and Molecular Dynamics guided by medium-resolution cryo-EM density maps can lead to highly accurate protein structure predictions. We continue to use medium to low resolution cryo-EM density maps in combination with other structural techniques, such as surface-induced dissociation, to improve protein structure elucidation.
Recent Related Publications:
Seffernick, J.T.; Canfield, S.M.; Harvey, S.R.; Wysocki, V.H.; Lindert, S. (2021), Prediction of Protein Complex Structure Using Surface-Induced Dissociation and Cryo-Electron Microscopy. Anal Chem 93 (21), 7596–7605.
Leelananda, S.P.; Lindert, S. (2020), Using NMR Chemical Shifts and Cryo-EM Density Restraints in Iterative Rosetta-MD Protein Structure Refinement. J Chem Inf Model 60 (5), 2522-2532.
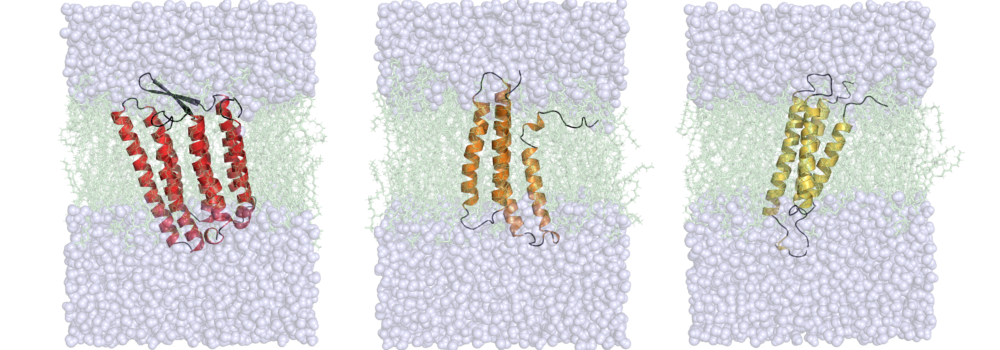
Leelananda, S.P.; Lindert, S. (2017), Iterative Molecular Dynamics-Rosetta Membrane Protein Structure Refinement Guided by Cryo-EM Densities. J Chem Theory Comput 13 (10), 5131-5145.
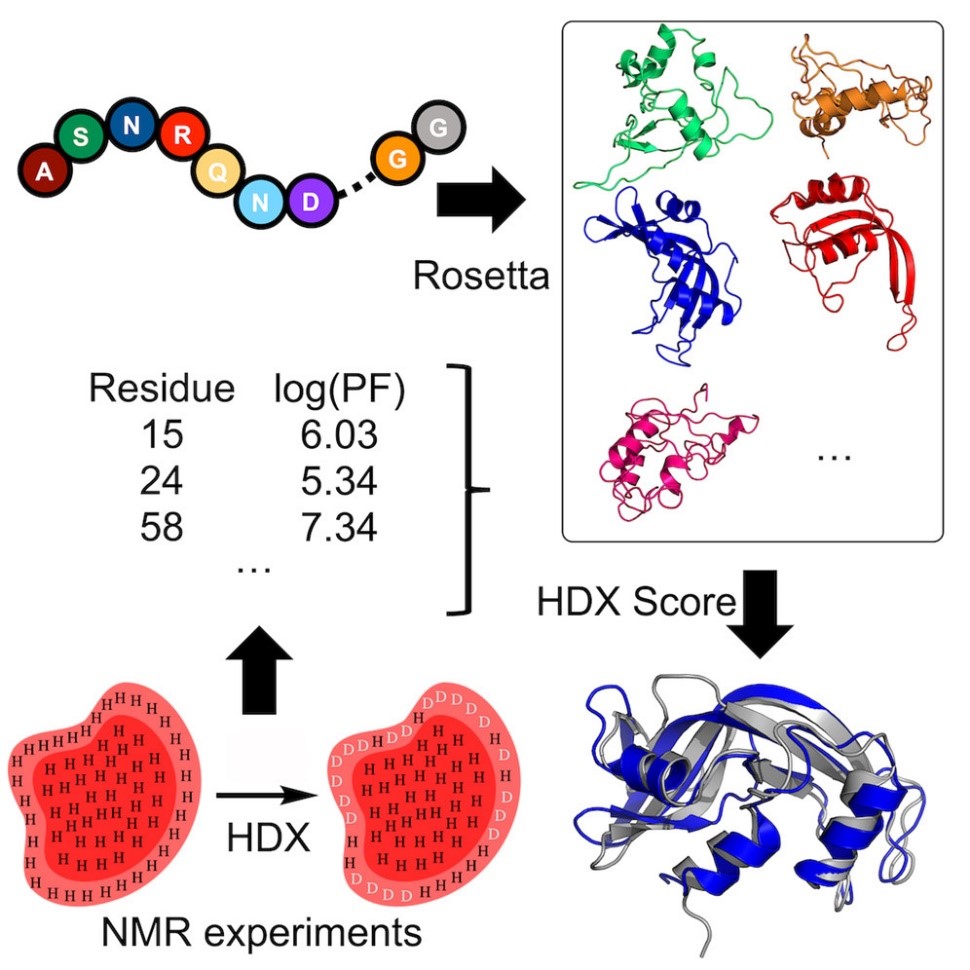
Nuclear Magnetic Resonance (NMR)
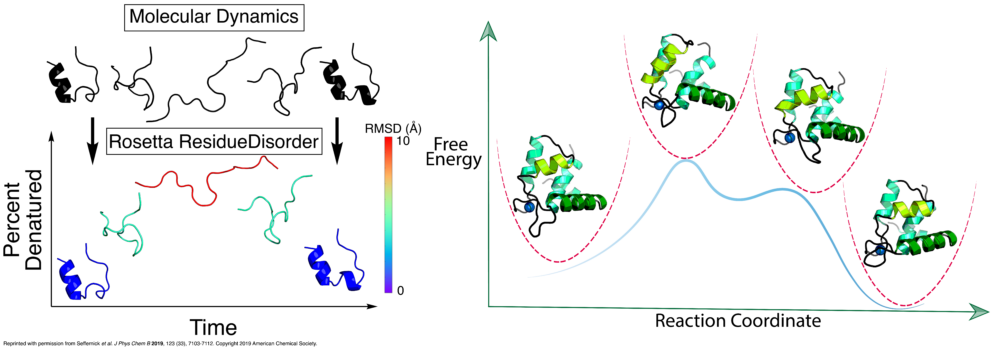
Hydrogen-deuterium exchange (HDX) measured by NMR provides protein structural information relating to residue solvent accessibility and flexibility. While this structural information is beneficial, it is insufficient to elucidate structures unambigiously. Thus, computational methods are needed to translate the HDX-NMR data into protein structures. We have developed a methodology to incorporate HDX-NMR data (categories of exchange rate and protection factors) into ab initio protein structure prediction using the Rosetta software to predict structures based on experimental agreement.
Recent Related Publications:
Nguyen, T.T.; Marzolf, D.R.; Seffernick, J.T.; Heinze, S.; Lindert, S. (2021), Protein structure prediction using residue-resolved protection factors from hydrogen-deuterium exchange NMR. Structure 30 (2), 313-320.
Marzolf, D.R.; Seffernick, J.T.; Lindert, S. (2021), Protein Structure Prediction from NMR Hydrogen-Deuterium Exchange Data. J Chem Theory Comput 17 (4), 2619-2629.
Leelananda, S.P.; Lindert, S. (2020), Using NMR Chemical Shifts and Cryo-EM Density Restraints in Iterative Rosetta-MD Protein Structure Refinement. J Chem Inf Model 60 (5), 2522-2532.
Simulations of Biomolecules
The lab is interested in understanding the relationship between dynamics, structure and function in proteins. For many biological systems the dynamics are crucial to their function. We use Molecular Dynamics (MD) simulations to investigate transitions between different conformational states, elucidate the dynamics of binding pockets (with implications for drug discovery) and gain insight into free energy barriers between states. Enhanced sampling methods, such as Gaussian accelerated MD (GaMD) can provide a means of extending the sampling of the conformational space beyond the current limits of nanosecond to microsecond timescales. Additionally, the lab makes use of various free energy methods such as umbrella sampling, thermodynamic integration, and adaptive steered molecular dynamics to quantitatively assess transitions between different thermodynamic states. The lab’s focus is on simulations of contraction in muscle cells. Both in cardiac and skeletal muscle, thin and thick filaments slide past each other to achieve muscle contraction. The process of contraction is regulated by calcium and encompasses multiple molecular events. Thus, our simulations target different proteins involved in contraction and also look at different scales (from a single protein to macromolecular complexes), with a particular focus on MD simulations of the cardiac troponin complex. The results of these simulations are then used to aid the identification of drugs targeting heart failure.
Troponin Reviews:
Bowman, J.D.; Lindert, S. (2019), Computational Studies of Cardiac and Skeletal Troponin. Front Mol Biosci 6:68.
Calcium Binding to Troponin C
Troponin’s role in regulating heart muscle contraction begins with Ca2+ binding to troponin C. We are focusing on computationally exploring the mechanism by which Ca2+ binding regulates heart muscle contraction. In our studies, we are applying molecular dynamics, thermodynamic integration, and adaptive steered MD simulations.
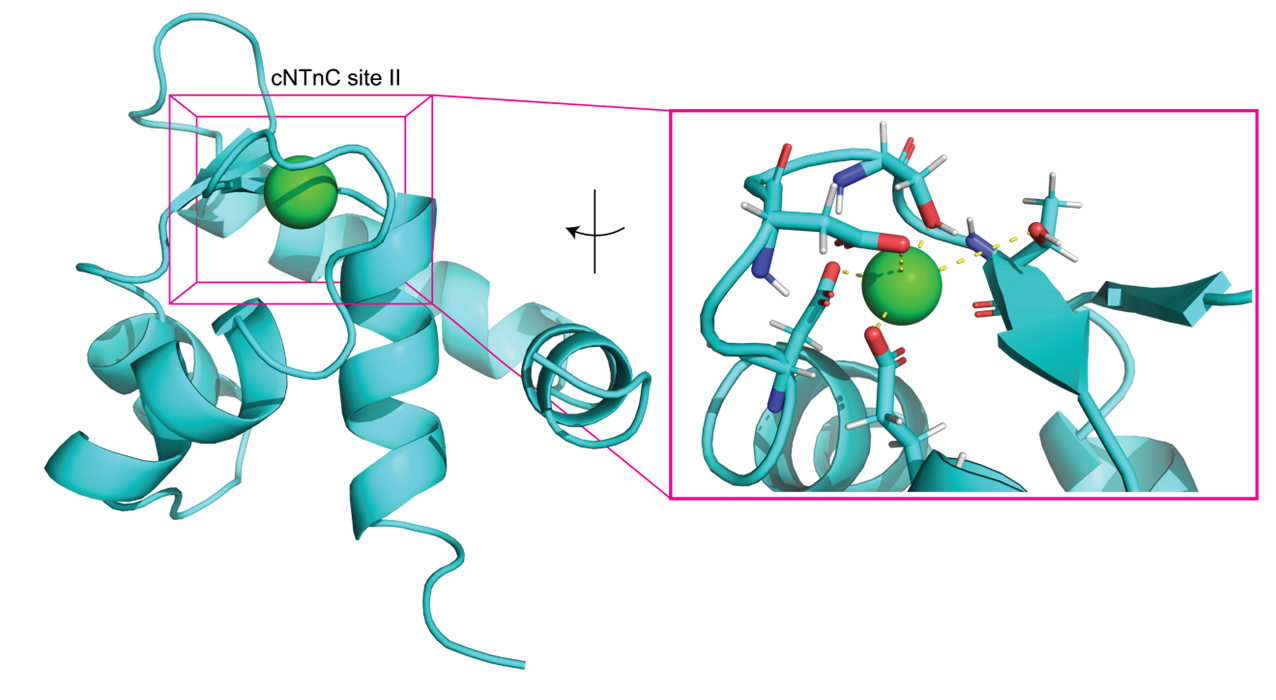 Recent Related Publications:
Recent Related Publications:
Hantz, E.R.; Lindert, S. (2022), Computational Exploration and Characterization of Potential Calcium Sensitizing Mutations in Cardiac Troponin C. J Chem Inf Model 62 (23), 6201-6208.
Hantz, E.R.; Lindert, S. (2021), Adaptive Steered Molecular Dynamics Study of Mutagenesis Effects on Calcium Affinity in the Regulatory Domain of Cardiac Troponin C. J Chem Inf Model 61 (6), 3052-3057.
Rayani, K.; Seffernick, J.T.; Li, A.Y.; Davis, J.P.; Spuches, A.M.; Van Petegem, F.; Solaro, R.J.; Lindert, S.; Tibbits, G.F. (2021), Binding of calcium and magnesium to human cardiac Troponin C. J Biol Chem 296, 100350.
Troponin C Hydrophobic Patch Opening
Ca2+ binding to troponin C initiates dynamic events where the N-terminal region of troponin C transitions from a “closed” to an “open” state. This transition reveals a hydrophobic patch that is able to bind the troponin I switch peptide region. We have studied this opening event extensively using molecular dynamics, steered MD, targeted MD, and umbrella sampling simulations.
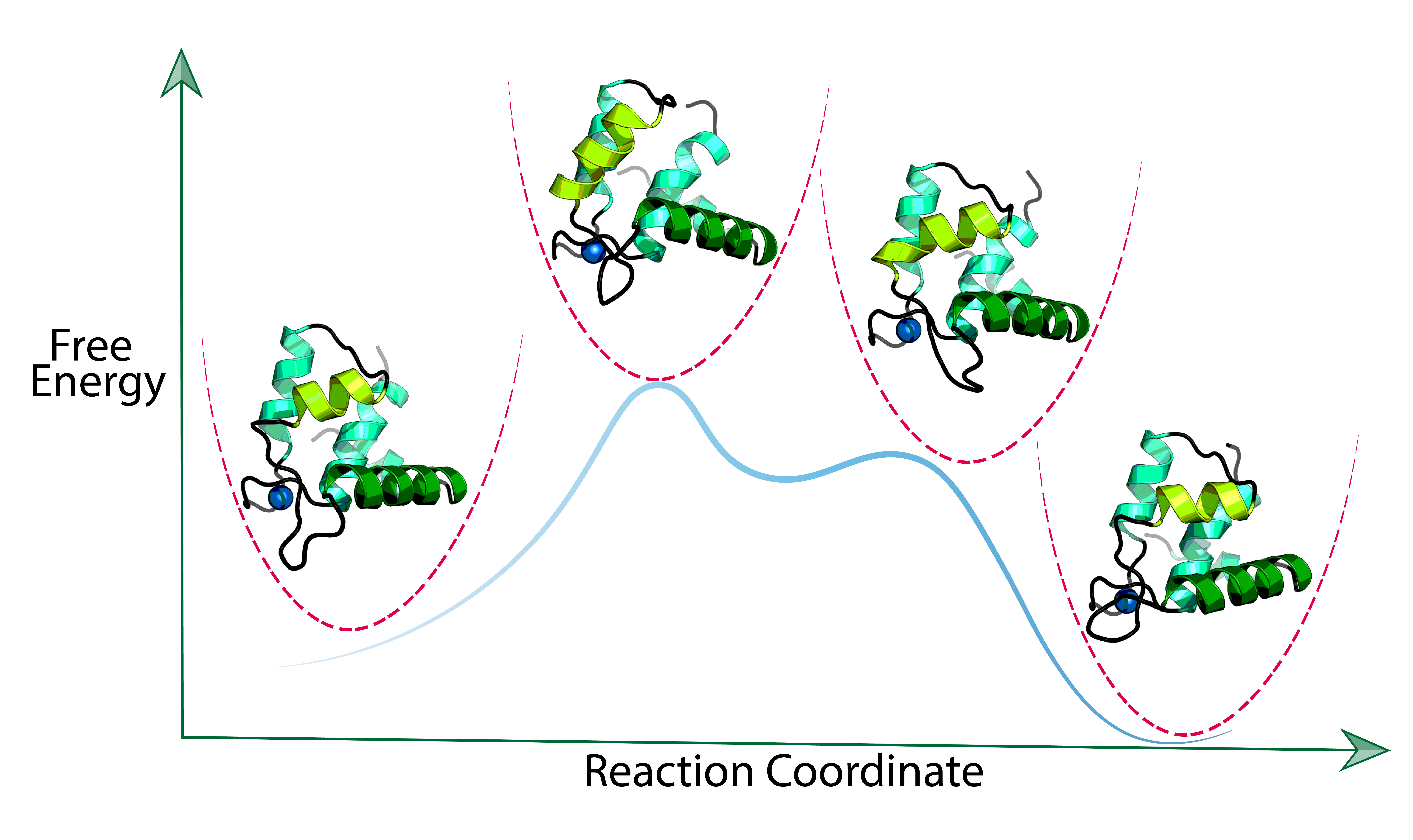
Recent Related Publications:
Cool, A.M.; Lindert, S. (2022), Umbrella Sampling Simulations Measure Switch Peptide Binding and Hydrophobic Patch Opening Free Energies in Cardiac Troponin. J Chem Inf Model 62 (22), 5666-5674.
Bowman, J.D.; Coldren, W.H.; Lindert, S. (2019), Mechanism of Cardiac Troponin C Calcium Sensitivity Modulation by Small Molecules Illuminated by Umbrella Sampling Simulations. J Chem Inf Model 59 (6), 2964-2972.
Bowman, J.D.; Lindert, S. (2018), Molecular Dynamics and Umbrella Sampling Simulations Elucidate Differences in Troponin C Isoform and Mutant Hydrophobic Patch Exposure. J Phys Chem B 122 (32), 7874–7883.
Troponin Dynamics
Troponin I blocks myosin binding heads from interacting with the thin filament. After Ca2+ binds to troponin C, a conformational shift occurs that removes troponin I from its blocking position to allow myosin heads to interact with actin, ultimately causing contraction to occur. In this state, troponin I binds to an “open” TnC. The transition between these two states and the effect of cardiomyopathic mutations on this transition is largely unexplored. Recently, we have begun researching this idea using conventional MD, accelerated MD, targeted MD, and umbrella sampling methods to quantify the impact of mutations on the dynamics and energetics of this transition.
Recent Related Publications:
Cool, A.M.; Lindert, S. (2023), Umbrella Sampling Simulations of Cardiac Thin Filament Reveal Thermodynamic Consequences of Troponin I Inhibitory Peptide Mutations. J Chem Inf Model. In Press. https://doi.org/10.1021/acs.jcim.3c00388
Cool, A.M.; Lindert, S. (2022), Umbrella Sampling Simulations Measure Switch Peptide Binding and Hydrophobic Patch Opening Free Energies in Cardiac Troponin. J Chem Inf Model 62 (22), 5666-5674.
Cool, A.M.; Lindert, S. (2021), Computational Methods Elucidate Consequences of Mutations and Post-translational Modifications on Troponin I Effective Concentration to Troponin C. J Phys Chem B 125 (27), 7388-7396.
Computer-Aided Drug Discovery (CADD)
Computational methods are a cornerstone of the early stages of the drug discovery pipeline. Structure-based drug discovery utilizes available structural data of known inhibitors and target receptor conformations obtained via structural biology techniques (NMR, X-ray crystallography, and cryo-EM). Structural information can also be supplemented with data from molecular dynamics simulations and structure prediction tools. Computational methods allow for virtual high throughput screenings of large compound libraries (106 – 109 compounds), where compounds are docked into the target receptor and the binding affinity is predicted. The lab works towards continuous improvements of computational methods for effective virtual screens and collaborates with experimental groups to test the best predicted compounds. We are interested in a variety of target systems: modulating calcium sensitivity of the thin filament with applications in treatment of heart failure, developing specific inhibitors for proteins critical to the pathology of various bacterial-based diseases, and developing inhibitors that selectively target cancer-related enzymes and receptors. The lab is also interested in incorporating machine learning/deep learning methods into all aspects of structure-based drug discovery.
CADD Reviews:
Turzo, S.B.A.; Hantz, E.R.; Lindert, S. (2022), Applications of machine learning in computer-aided drug discovery. QRB Discov 3, e14.
Leelananda, S.P.; Lindert, S. (2016), Computational methods in drug discovery. Beilstein J Org Chem 12, 2694–2718.
Congestive Heart Failure
Cardiac muscle contraction is a calcium-dependent mechanism which is regulated by the troponin protein complex (cTn) and specifically by the N-terminal domain of its calcium binding subunit (cNTnC). There is an increasing need for the development of small molecules that increase calcium sensitivity without altering systolic calcium concentration, thereby strengthening cardiac function. Our group takes a structure-based drug discovery and cheminformatics approach to identify and optimize such small molecules in collaboration with experimentalists for in vitro and in vivo testing. This work is in collaboration with the Davis and Reiser labs.
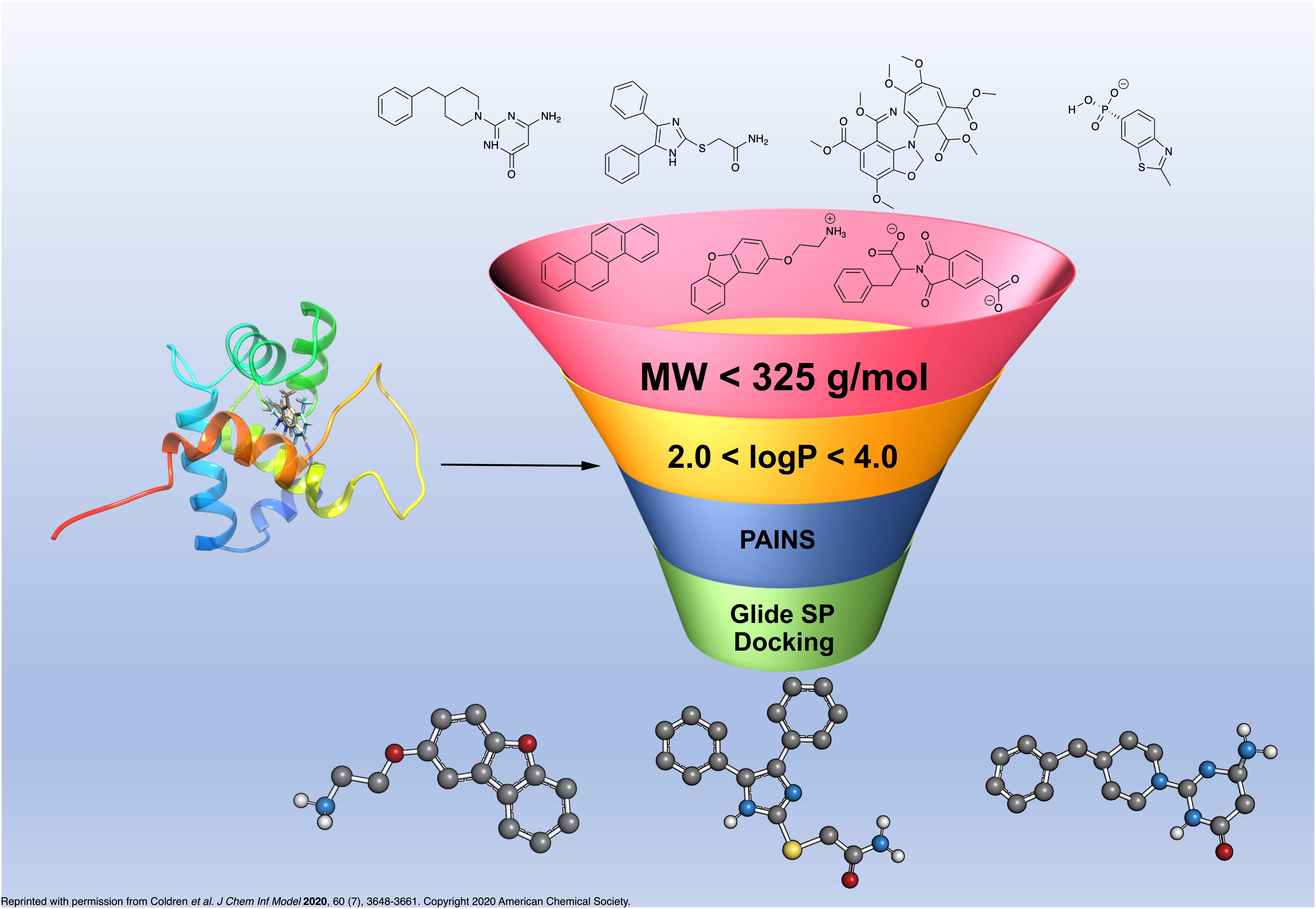
Recent Relevant Publications:
Hantz, E.R.; Tikunova, S.B.; Belevych, N.; Davis, J.P.; Reiser, P.J.; Lindert, S. (2023), Targeting Troponin C with Small Molecules Containing Diphenyl Moieties: Calcium Sensitivity Effects on Striated Muscles and Structure–Activity Relationship. J Chem Inf Model. In Press. DOI:10.1021/acs.jcim.3c00196
Coldren, W.H.; Tikunova, S.B.; Davis, J.P.; Lindert, S. (2020), Discovery of Novel Small-Molecule Calcium Sensitizers for Cardiac Troponin C: A Combined Virtual and Experimental Screening Approach. J Chem Inf Model 60 (7), 3648-3661.
Aprahamian, M.L.; Tikunova, S.B.; Price, M.V.; Cuesta, A.F.; Davis, J.P.; Lindert, S. (2017), Successful Identification of Cardiac Troponin Calcium Sensitizers Using a Combination of Virtual Screening and ROC Analysis of Known Troponin C Binders. J Chem Inf Model 57 (12), 3056-3069.
Bacterial Disease Targets
Salmonella is a common bacterial disease that affects the intestinal tract, for which there are currently no vaccines or therapeutics for humans. We aim to develop target specific inhibitors for various enzymes that are critical for Salmonella’s metabolism. We utilize high-throughput virtual screening and lead optimization techniques. This work is in collaboration with the Gopalan lab.
Recent Relevant Publications:
Thirugnanasambantham, P.; Kovvali, S.; Cool, A.M.; Gao, Y.; Sabag-Daigle, A.; Boulanger, E.F.; Mitton-Fry, M.J.; Capua, A.D.; Behrman, E.J.; Wysocki, V.H.; Lindert, S.; Ahmer, B.M.M.; Gopalan, V. (2022), Serendipitous Discovery of a Competitive Inhibitor of FraB, a Salmonella Deglycase and Drug Target. Pathogens 11 (10), 1102.
Sengupta, A.; Wu, J.; Seffernick, J.T.; Sabag-Daigle, A.; Thomsen, N.; Chen, T.H.; Di Capua, A.; Bell, C.E.; Ahmer, B.M.M.; Lindert, S.; Wysocki, V.H.; Gopalan, V. (2019), Integrated use of biochemical, native mass spectrometry, computational and genome-editing methods to elucidate the mechanism of a Salmonella deglycase. J Mol Biol 431 (22), 4497-4513.
With the recent increase of antibiotics use, bacteria have begun to develop multidrug resistance. Therefore, a critical need for new antibiotics to treat these pathogens has arisen. In collaboration with Dr. Mitton-Fry, we aim to identify and optimize novel bacterial topoisomerase and DNA gyrase inhibitors for gram-positive bacteria.
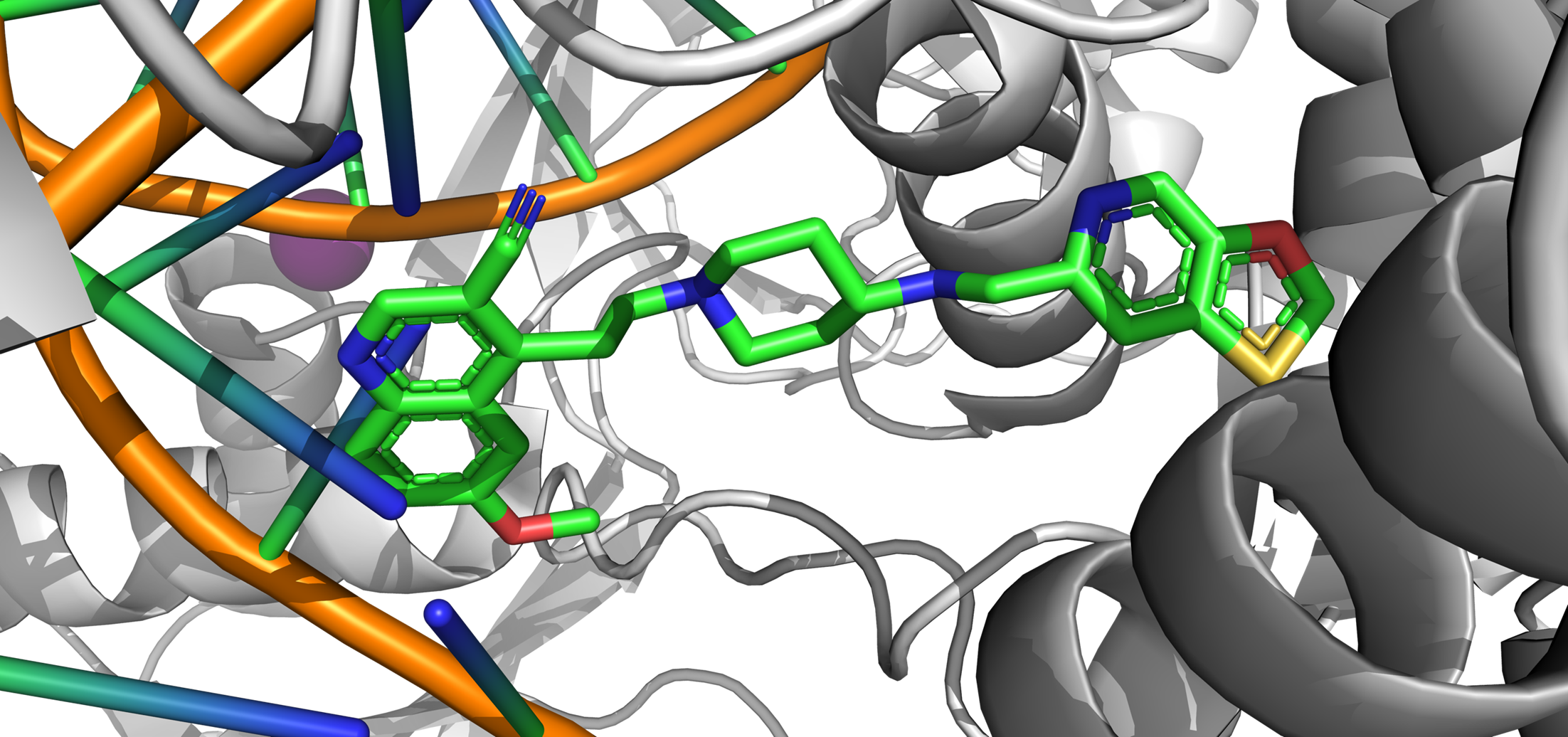
Recent Relevant Publications:
Lu, Y.; Vibhute, S.; Li, L.; Okumu, A.; Ratigan, S.C.; Nolan, S.; Papa, J.L.; Mann, C.A.; English, A.; Chen, A.; Seffernick, J.T.; Koci, B.; Duncan, L.R.; Roth, B.; Cummings, J.E.; Slayden, R.A.; Lindert, S.; McElroy, C.A.; Wozniak, D.J.; Yalowich, J.; Mitton-Fry, M.J. (2021), Optimization of TopoIV Potency, ADMET Properties, and hERG Inhibition of 5-Amino-1,3-dioxane-Linked Novel Bacterial Topoisomerase Inhibitors: Identification of a Lead with In Vivo Efficacy against MRSA. J Med Chem 64 (20), 15214-15249.
Lu, Y.; Papa, J.L.; Nolan, S.; English, A.; Seffernick, J.T.; Shkolnikov, N.; Powell, J.; Lindert, S.; Wozniak, D.J.; Yalowich, J.; Mitton-Fry, M.J. (2020), Dioxane-Linked Amide Derivatives as Novel Bacterial Topoisomerase Inhibitors against Gram-Positive Staphylococcus aureus. ACS Med Chem Lett 11 (12), 2446-2454.
Li, L.; Okumu, A.A.; Nolan, S.; English, A.; Vibhute, S.; Lu, Y.; Hervert-Thomas, K.; Seffernick, J.T.; Azap, L.; Cole, S.L.; Shinabarger, D.; Koeth, L.; Lindert, S.; Yalowich, J.; Wozniak, D.J.; Mitton-Fry, M.J. (2019), 1,3-Dioxane-linked Bacterial Topoisomerase Inhibitors with Enhanced Antibacterial Activity and Reduced hERG Inhibition. ACS Infect Dis 5 (7), 1115-1128.
Cancer Related Targets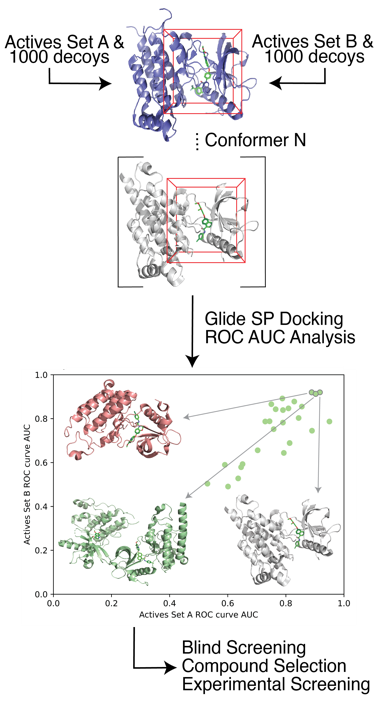 Recently, we have developed a protocol for target receptor conformation selection which led to an improvement in the true positive prediction rate of structure-based drug design. We showed that our methods are generalizable to a wide array of target systems. Additionally, for a subset of five cancer-related kinases we validated our virtual screening approach with experimental testing, which led to the identification of 22 potent kinase inhibitors ranging from low affinity.
Recently, we have developed a protocol for target receptor conformation selection which led to an improvement in the true positive prediction rate of structure-based drug design. We showed that our methods are generalizable to a wide array of target systems. Additionally, for a subset of five cancer-related kinases we validated our virtual screening approach with experimental testing, which led to the identification of 22 potent kinase inhibitors ranging from low affinity.
Recent Relevant Publications:
Hantz, E.R.; Lindert, S. (2022), Actives-Based Receptor Selection Strongly Increases the Success Rate in Structure-Based Drug Design and Leads to Identification of 22 Potent Cancer Inhibitors. J Chem Inf Model 62 (22), 5675-5687.