
Research Areas
Our lab’s research focuses on the characterization and understanding of the dynamics, interactions, and function of proteins at the atomistic level by nuclear magnetic resonance (NMR) spectroscopy in solution and computational modeling. We are also developing new NMR methods, databases, and computational tools for the analysis of complex mixtures for application in metabolomics. Such tools are provided to the public in the form of software and web servers. Our research involves wetlab biochemistry, experimental NMR, high-performance computation, and other biophysical techniques. We are primarily supported by the National Science Foundation and the National Institutes of Health.
Functional Protein Dynamics by NMR Spectroscopy
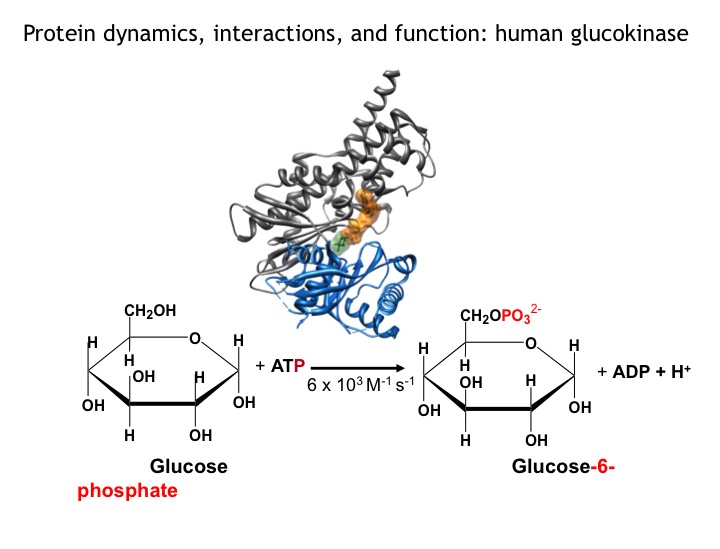
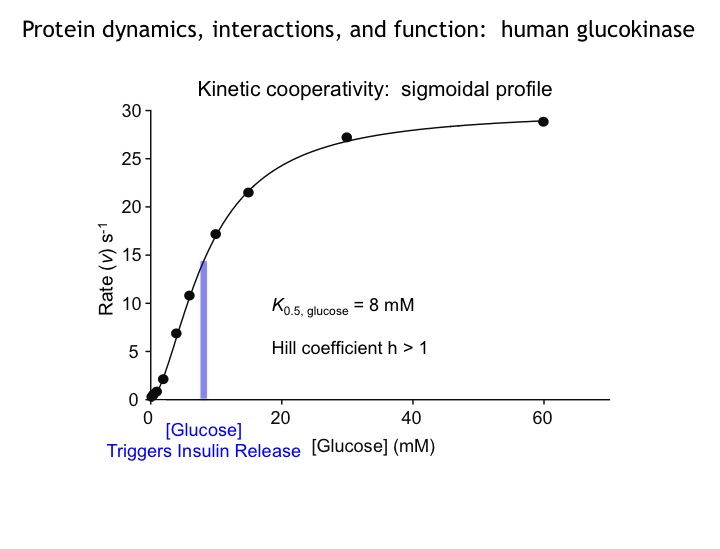
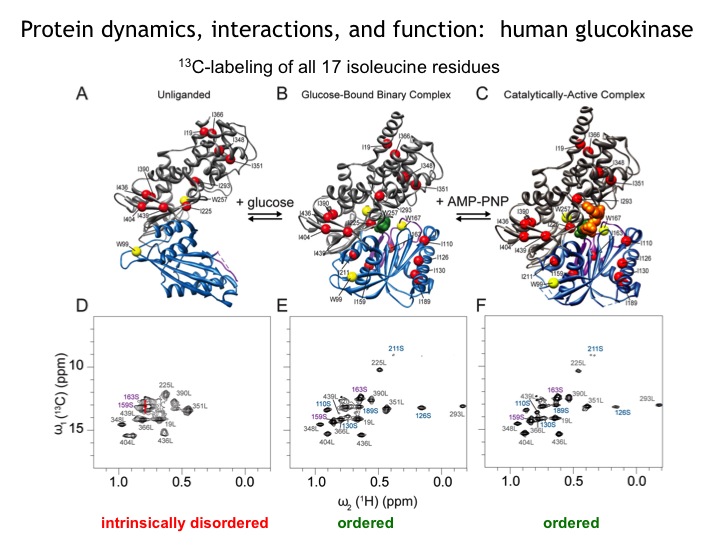
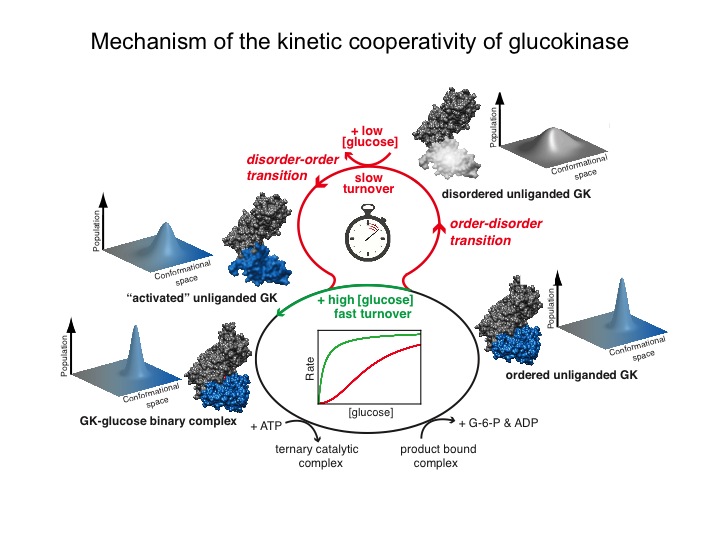
Many protein systems show cooperative behavior, which permits them to sensitively respond to changes of their environment. For example, Glucokinase (GCK) is a key metabolic enzyme that functions as the body’s principal glucose sensor. GCK catalyzes the first and rate-limiting step of glycolysis, which is the phosophorylation of glucose. This process regulates the rate at which insulin is secreted by the pancreas by using a cooperative kinetic response to increasing glucose concentrations. GCK dysfunction can lead to severe diseases, such maturity onset diabetes of the young type II (MODY-II) and persistent hypoglycemic hyperinsulinemia of infancy (PHHI). GCK, which is a 52 kDa monomeric enzyme, displays a characteristic kinetic cooperativity profile, whereby its sigmoidal shape markedly differs from classical Michaelis-Menten kinetics. In collaboration with Prof. Brian Miller, we study the origin of this effect at atomistic detail. For this purpose we specifically label all 17 isoleucine residues of GCK with carbon-13 in their side chains (Cd1 positions) and monitor by NMR the behavior of the different regions that carry the isoleucine labels as this enzymes moves along its reaction coordinate. The NMR results reveal that in its unliganded state GCK is an intrinsically disordered enzyme containing a disordered region in its small domain (blue) and it undergoes conformational exchange on the millisecond time scale between the active and a less active state. The population of the less active state increases as the glucose concentration decreases, leading to the characteristic drop in the middle of the sigmoidal kinetic profile. The mechanism can be depicted by the time-delay loop, which is entered upon the order-disorder transition of the small domain. Spontaneous and ligand-induced ordering brings GKC back to the high turnover regime.
Other proteins that we study in our lab are arginine kinase (in collaboration with Prof. Chapman, OHSU), the sodium-calcium exchanger (NCX, in collaboration with Prof. Salinas, U. Sao Paulo), Copper-ATPase and sodium-potassium-ATPase, the interaction modes of the N-terminal domain of p53 with MDM2/MDMX.
top
Predictive Understanding of Protein Behavior by the Combination of molecular dynamics computer simulations and NMR
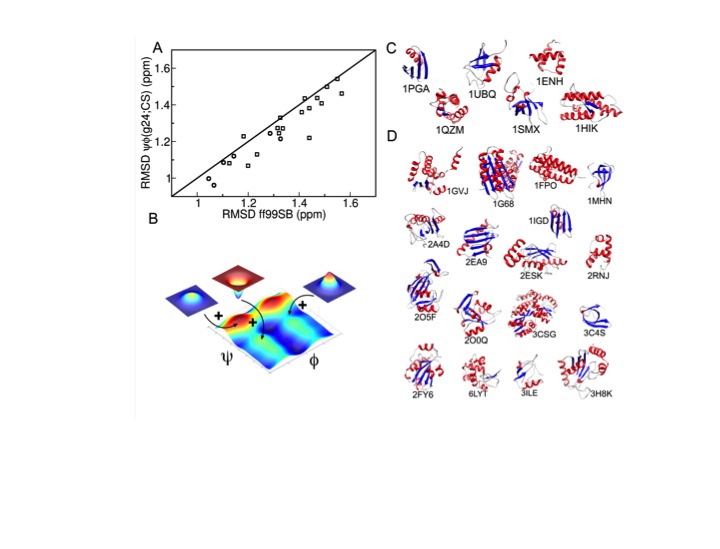
NMR spectroscopy provides a wealth of information about proteins at atomic detail. This information can be significantly enhanced by the comparison experimental data with in silico studies of the same system. In our lab, we are using primarily long-term molecular dynamics simulations (MD), performed on the Ohio supercomputer, which is located on West campus. All-atom protein simulations in explicit solvent now routinely extend into the microsecond range and beyond. We develop methods for the computation of different types of NMR parameters from such simulations, including spin relaxation parameters (S2 order parameters), scalar J-couplings, and residual dipolar couplings (RDCs). The quantitative comparison between computed and experimental NMR parameters permits the detailed assessment of the quality and accuracy of the computer models.
NMR chemical shifts belong to the most abundant NMR parameters, but because of their quantum-chemical origin their interpretation in terms of protein structure and dynamics is challenging. We are developing and applying the PPM software for the back-calculation of chemical shifts from large conformational ensembles produced by MD simulations.
Conversely, we are developing protocols for the improvement of MD simulations so that they become increasingly quantitative even for systems for which no NMR data are available. For this purpose, we use experimental NMR chemical shifts and residual dipolar couplings of full-length proteins and derived the ff99SBnmr1 and the ff99SB_ΦΨ(g24;CS) force fields, which prove to be of high quality and they are well transferable between different protein systems.
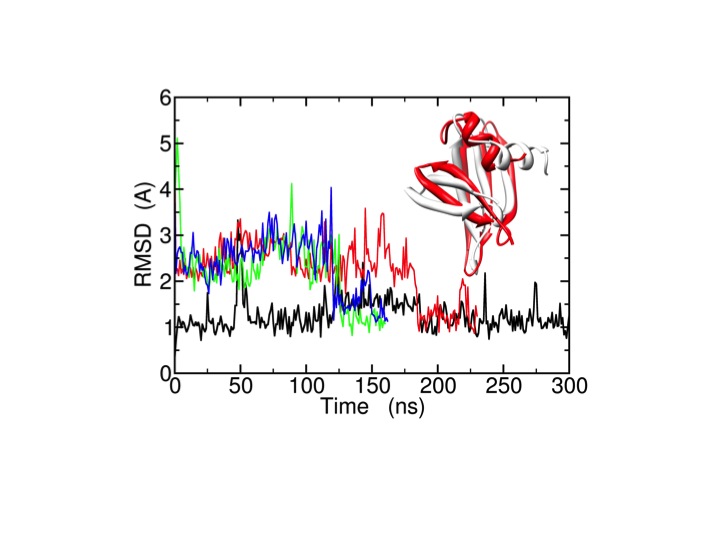
These efforts aim at an increasingly predictive understanding of proteins, their properties, and function. As an example, we apply microsecond all-atom molecular dynamics simulations using ff99SB_ΦΨ(g24;CS) for the improvement of the quality of experimental low-resolution protein structures and structural models (decoys) generated by protein structure prediction methods (e.g. Rosetta). For the protein, long MD simulations spontaneously bring the wrong decoys to the native structure (X-ray crystal structure).
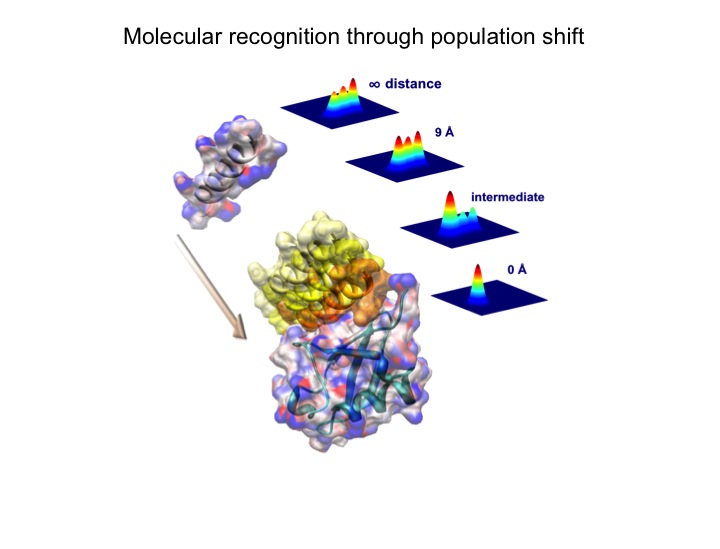
Elucidation of the mechanism of biomacromolecular recognition events is a topic of intense interest. The recognition dynamics of Ubiquitin can be studied via microsecond all-atom molecular dynamics simulations providing both thermodynamic and kinetic information. The high level of consistency found with respect to experimental NMR data lends support to the accuracy of the in silico representation of the conformational substates and their interconversions in free ubiquitin. Using an energy-based reweighting approach, the statistical distribution of conformational states of ubiquitin can be monitored, for example, as a function of the distance between ubiquitin and its binding partner Hrs-UIM suggesting a significant population shift in the thermal distribution of backbone conformers of Hrs-UIM during this protein-protein docking event.
Related tools are explored in our lab for the understanding of allostery during molecular recognition events, which is the propagation of structural and dynamic changes over extended distances in proteins.
Together, the exploration of synergies between experiment and simulations provides an increasingly predictive understanding of protein properties and function.
top
New NMR methods for resolution and sensitivity enhancement
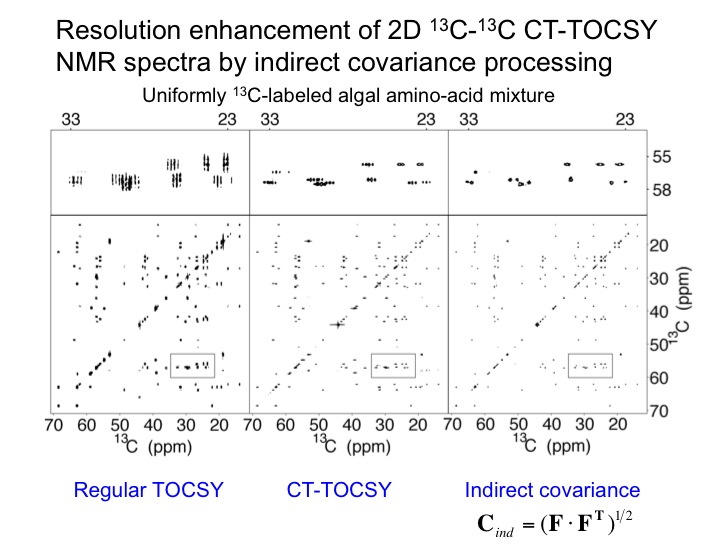
The analysis of multidimensional NMR spectra benefits from high sensivitiy and high spectral resolution. We are developing new NMR methods toward this goal as expemplified with a 2D TOCSY spectrum of a uniformly 13C-labeled algal amino-acid mixture. The application of constant-time TOCSY together with indirect covariance processing provides a substantial improvement in spectral resolution, which permits the unambiguous identification of NMR cross-peaks in crowded regions of the NMR spectrum. We are developing this and other tools for the semi-automated analysis of 2D NMR spectra of complex mixtures as encountered in many types of applications of metabolomics.
top
From large to small molecules: complex biological mixture analysis and function with application to metabolomics
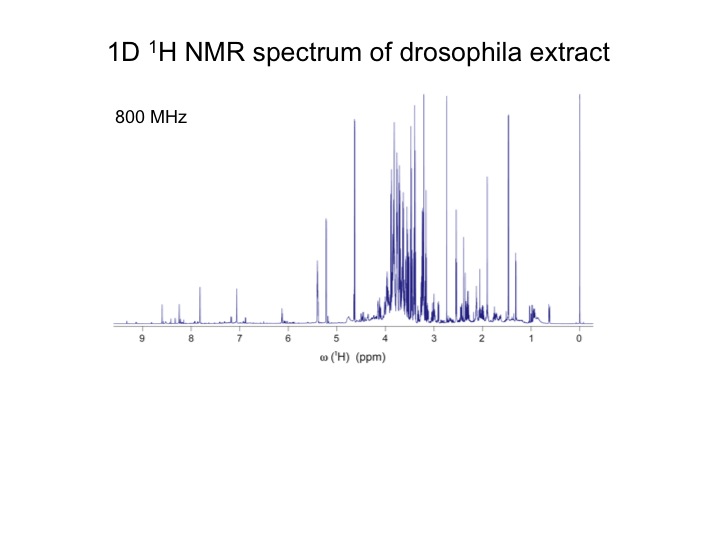
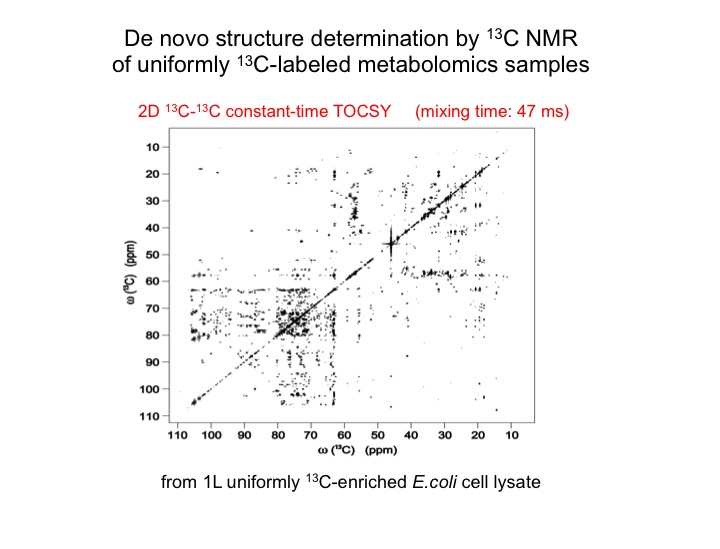
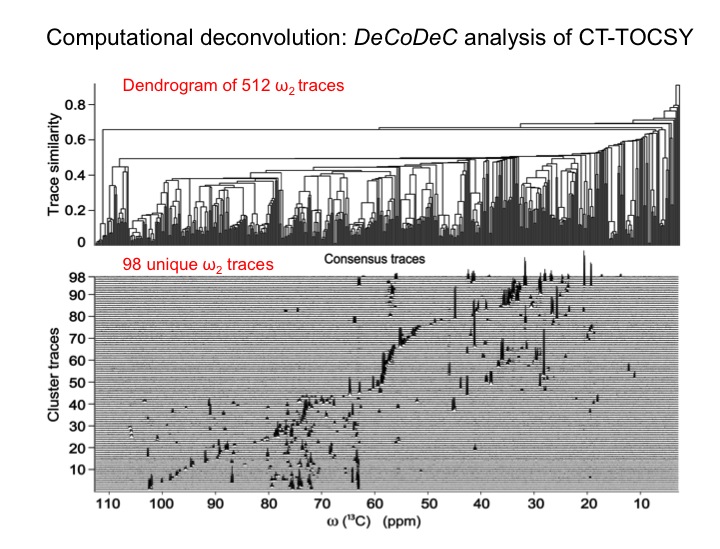


The analysis of complex biological mixtures by NMR without the need of extensive purification is an important objective. Since NMR spectra of metabolite mixtures are often inordinately complex, new methods are required that provide sufficient resolution.
The metabolic makeup of a biological system (cell, tissue, biofluid, etc) is a key determinant of its biological state. Comprehensive identification and accurate quantification of the metabolites of such systems form two of the most critical components of metabolomics. NMR spectroscopy is a unique tool for this purpose providing a wealth of atomic-detail information without requiring extensive fractionation of samples. Two of the bottlenecks in metabolite identification are the incompleteness and inaccuracy of metabolite databanks. We are working on two complementary approaches to overcome these problems. Since many metabolites are not present in metabolite databanks, the first approach extracts their carbon backbone structures (topology), which is a prerequiste for de novo structure determination. From a single sample of E. coli, this allows, for example, the determination of up to 112 topologies of unique metabolites that constitute the “topolome” of a cell.
Since databanks are designed to query complete 1D NMR spectra, the querying of TOCSY traces against 1D NMR spectra in databanks often results in imperfect matches. To overcome this issue we are assembling a customized 13C TOCSY database, which substantially improves the accuracy of database query of 13C TOCSY traces. Together these new tools open up the prospect to routinely yet accurately analyze an increasingly complex and diverse range of complex mixtures with many applications to metabolomics.