

Accurate Quantum Chemistry in Many Electronic States
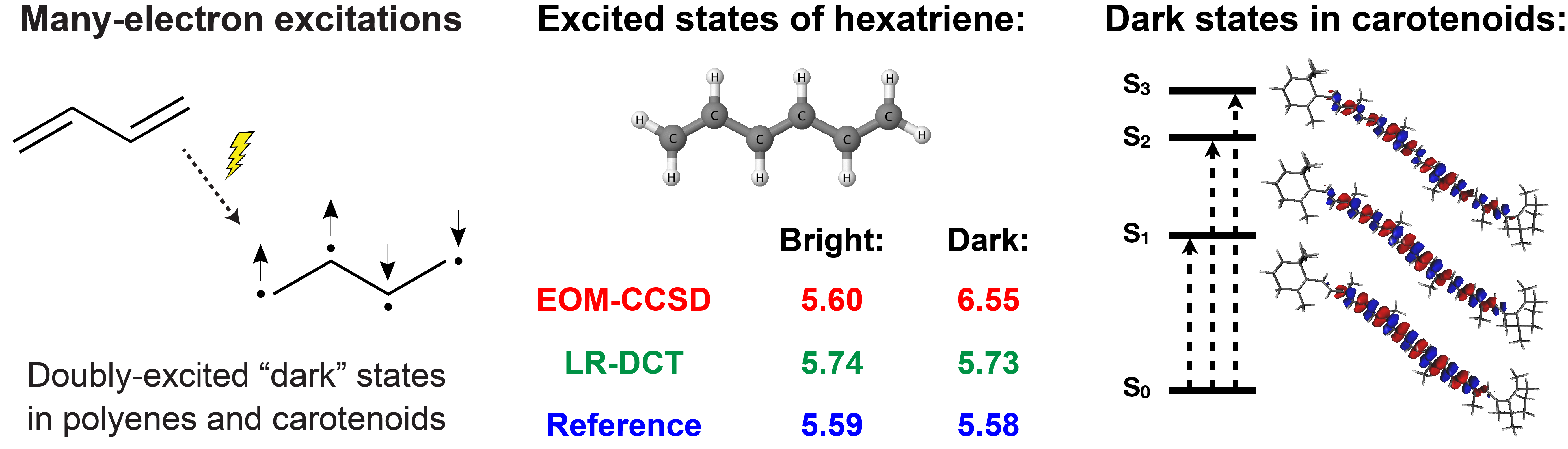
Research in our group aims to develop new theoretical methods for the simulations of light-induced and non-equilibrium processes in chemical systems with complex electronic structure. Our specific focus is the development of first-principles electronic structure approaches that can efficiently describe electron correlation effects and charge/energy transfer in many (10’s or even 100’s) electronic states.
Reliable Theories for Spectroscopic Properties of Strongly Correlated Systems
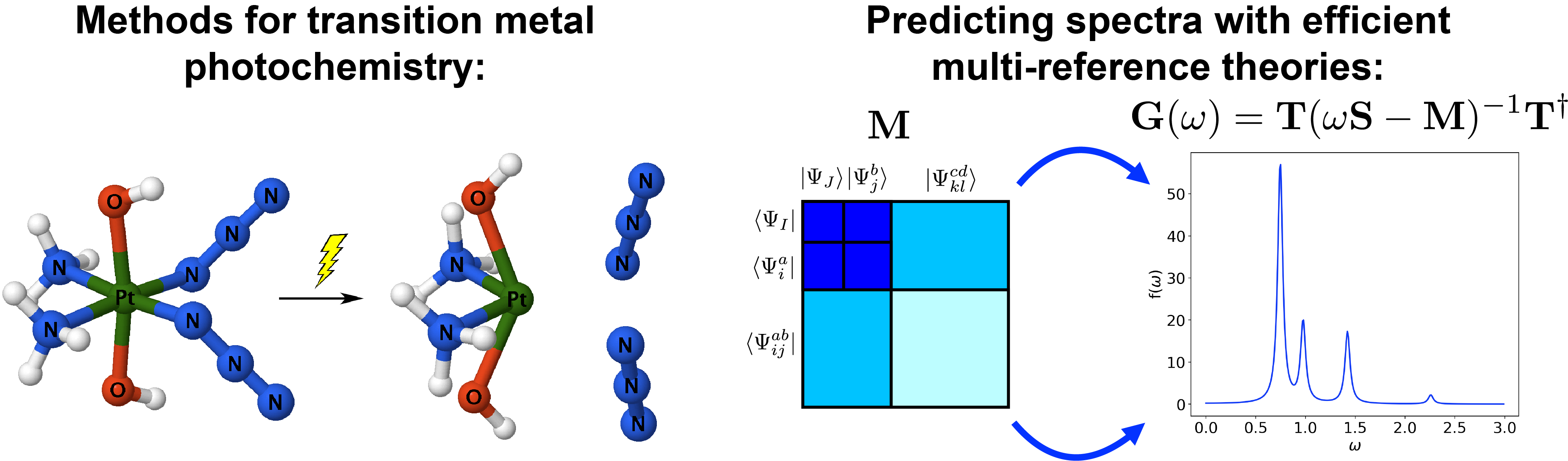
Our group is developing new theoretical approaches that can reliably simulate spectroscopic properties of strongly correlated systems. Strong electron correlation originates due to significant mixing of degenerate (or near-degenerate) electronic configurations and is very common in chemistry. We are working on the development of methods that can reliably describe effects of strong correlation, provide direct access to important spectroscopic properties, and, yet, are computationally affordable in large systems (such as transition metal compounds).
Methods for X-ray Spectroscopies and High-Energy Processes
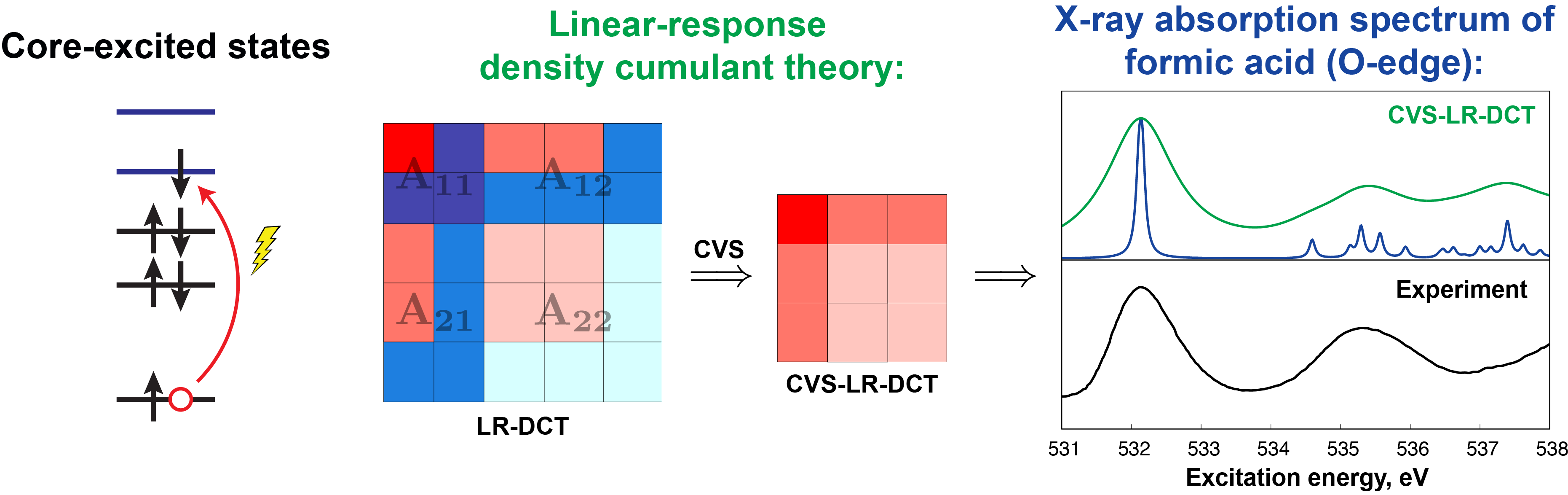
Computations of core-level excitations are very challenging as they require simulating excited states selectively in the high-energy spectral region and a balanced treatment of electron correlation, orbital relaxation, and relativistic effects, often combined with large uncontracted basis sets. We are developing new techniques that incorporate accurate description of electron correlation into efficient simulations of X-ray spectra.
Quantum Chemistry Software Development

All methods developed in our group are implemented in Prism, a standalone open-source Python program for excited-state and spectroscopic simulations of molecules. Prism is interfaced with popular quantum chemistry packages, such as Psi4 and Pyscf.