In this project, we integrate the drug discovery and delivery technologies of projects I and II to design cell-permeable biomolecules as intracellular biologics and chemical probes, in three different approaches. In the first approach, we generate intracellular biologics by conjugating cyclic CPPs to peptides, proteins, nucleic acids, and protein-nucleic acid complexes to render the latter cell permeable and biologically active. Published examples include potent inhibitors against MDM2 [1], b-catenin [1], calcineurin [2], protein-tyrosine phosphatases 1B [3], and bacterial virulence factor [4]. In our second approach, monocyclic and bicyclic peptide libraries are designed to integrate target-binding and cell-penetrating moieties into the same macrocyclic peptide. Screening of the libraries results in macrocyclic peptide ligands that are cell-permeable and biologically active against intracellular proteins. By using this approach, we have discovered potent, specific, cell-permeable, and metabolically stable inhibitors against some of the most challenging PPI targets including Ras [5-7], CAL PDZ domain [8], and NEMO [9] (Figure 1). In the third approach, we have engineered a family of membrane translocation domains (MTDs), which can deliver essentially any protein or other biomolecules into mammalian and plant cells.
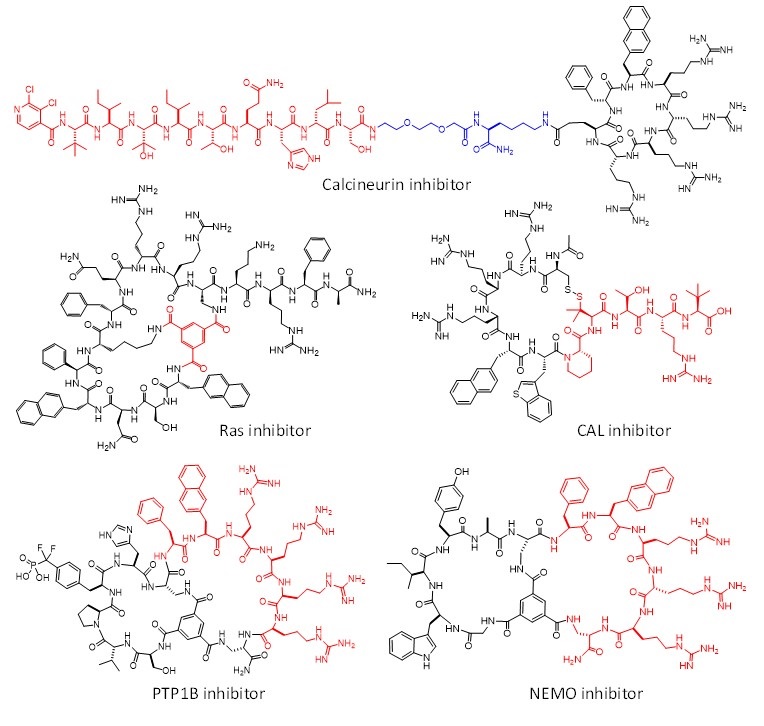
Figure 1. Examples of cell-permeable macrocyclic peptidyl inhibitors against intracellular proteins.
Current and future studies on this project include:
-
Optimization of peptidyl inhibitors against b-catenin, calcineurin, K-Ras, and PDZ domains
-
Development of macrocyclic peptidyl inhibitors against novel targets
-
Development of cell-permeable proteins as PPI inhibitors
-
Targeted protein degradation (PROTACs) with cell-permeable peptides and proteins
Key Publications:
- Dougherty, P. G., Wen, J., Pan, X., Koley, A., Ren, J.-G., Sahni, A., Basu, R., Salim, H., Appiah Kubi, G., Qian, Z., and Pei, D. (2019) Enhancing the Cell-Permeability of Stapled Peptides with a Cyclic Cell-Penetrating Peptide. Med. Chem. 62, 10098-10107.
- Dougherty, P. G., Karpurapu, M., Koley, A., Lukowski, J. K., Qian, Z., Srinivas Nirujogi, T., Rusu, L., Chung, S., Hummon, A. B., Li, H. W., Christman, J. W., and Pei, D. (2020) A Peptidyl Inhibitor that Blocks Calcineurin-NFAT Interaction and Prevents Acute Lung Injury. Med. Chem. 63, 12853–12872.
- Lian, W., Jiang, B., Qian, Z., and Pei, D. (2014) Cell-Permeable Bicyclic Peptide Inhibitors against Intracellular Proteins. Am. Chem. Soc. 136, 9830-9833.
- Zhang, W., Lin, M., Yan, Q., Budachetri, K., Hou, L., Sahni, A., Liu, H., Han, N.-C., Lakritz, J., Pei, D., and Rikihisa, Y. (2021) An intracellular nanobody targeting T4SS effector inhibits Ehrlichia Proc. Natl. Acad. Sci. U. S. A. 118(18) e2024102118.
- Upadhyaya, P., Qian, Z., Selner, N. G., Clippinger, S. R., Wu, Z., Briesewitz, R., and Pei, D. (2015) Inhibition of Ras signaling by blocking Ras-effector interactions with cyclic peptides. Chem. Int. Ed. 54, 7602-7606.
- Trinh, T. B., Upadhyaya, P., Qian, Z., and Pei, D. (2016) Discovery of a Direct Ras Inhibitor by Screening a Combinatorial Library of Cell-Permeable Bicyclic Peptides. ACS Comb Sci. 18, 75-85.
- Buyanova, M., Cai, S., Cooper, J., Rhodes, C., Salim, H., Sahni, A., Upadhyaya, P., Yang, R., Sarkar, A., Li, N., Wang, Q. E., & Pei, D. (2021) Discovery of a Bicyclic Peptidyl Pan-Ras Inhibitor. Med. Chem. 64, 13038–13053.
- Dougherty, P. G., Wellmerling, J. H., Koley, A., Lukowski, J. K., Hummon, A. B., Cormet-Boyaka, E., and Pei, D. (2020) Cyclic Peptidyl Inhibitors against CAL-CFTR Interaction for Treatment of Cystic Fibrosis. Med. Chem. 63, 15773–15784.
- Rhodes, C. A., Dougherty, P. G., Cooper, J., Qian, Z., Lindert, S., Wang, Q.-E., and Pei, D. (2018) Cell-Permeable Bicyclic Peptidyl Inhibitors against NEMO-IkB Kinase Interaction Directly from a Combinatorial Library. Am. Chem. Soc. 140, 12102-12110.