155. Discovery of a Cyclic Cell-Penetrating Peptide with Improved Endosomal Escape and Cytosolic Delivery Efficiency
Marina Buyanova, Ashweta Sahni, Rui Yang, Amar Sarkar, Heba Salim, and Dehua Pei, : Mol. Pharmaceutics 2022, 19, 1378−1388
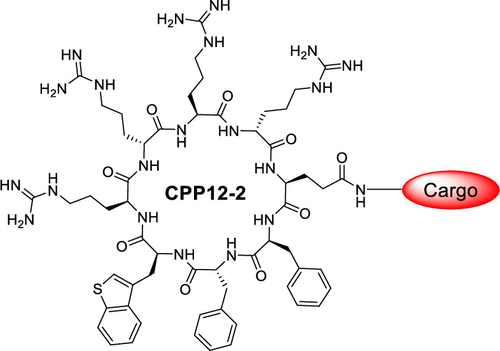
Cyclic cell-penetrating peptide 12 (CPP12) is highly efficient for the cytosolic delivery of a variety of cargo molecules into mammalian cells in vitro and in vivo. However, its cytosolic entry efficiency is substantially reduced at lower concentrations or in the presence of serum proteins. In this study, CPP12 analogs were prepared by replacing its hydrophobic residues with amino acids of varying hydrophobicity and evaluated for cellular entry. Substitution of L -3-benzothienylalanine (Bta) for L -2-naphthylalanine (Nal) resulted in CPP12-2, which exhibits up to 3.8-fold higher cytosolic entry efficiency than CPP12, especially at low CPP concentrations; thanks to improved endosomal escapeefficiency. CPP12-2 is well suited for the cytosolic delivery of highly potent cargos to achieve biological activity at low concentrations.
154. How Do Biomolecules Cross the Cell Membrane?
Dehua Pei, Acc. Chem. Res. 2022, 55, 3, 309–318, 10.1021/acs.accounts.1c00560
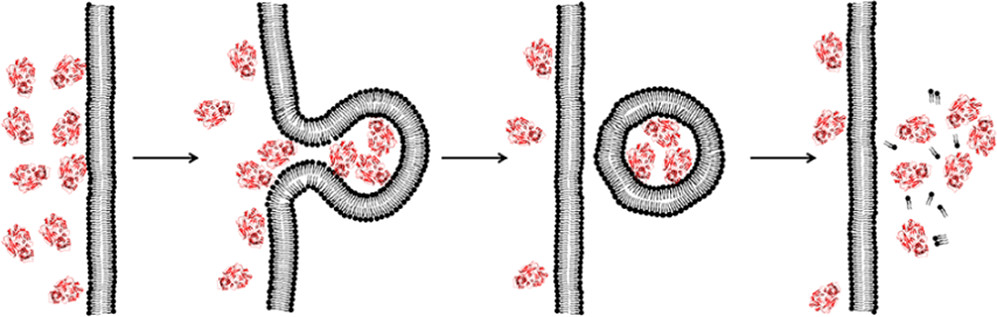
In this Account, I introduce a previously unrecognized, fundamental membrane translocation mechanism which we have termed the vesicle budding-and-collapse (VBC) mechanism. Through VBC, biomolecules of diverse sizes and physicochemical properties autonomously translocate across cell membranes topologically (i.e., from one side to the other side of the membrane) but not physically (i.e., without going through the membrane). We have demonstrated that CPPs and bacterial protein toxins escape the endosome by the VBC mechanism in giant unilamellar vesicles as well as live mammalian cells. This advance resulted from studies in which we labeled the biomolecules with a pH-sensitive, red-colored dye (pHAb) and phosphatidylserine with a pH-insensitive green dye (TopFluor) and monitored the intracellular trafficking of the biomolecules in real time by confocal microscopy. In addition, by enlarging the endosomes with a kinase inhibitor, we were able to visualize the structural changes of the endosomes (i.e., endosomal escape intermediates) as they went through the VBC process. I postulate that bacterial/viral/eukaryotic proteins, nonenveloped viruses, and synthetic drug delivery vehicles (e.g., polyplexes, lipoplexes, and lipid nanoparticles) may also escape the endosome by inducing VBC. Furthermore, I propose that VBC may be the mechanism that drives the bacterial TAT and eukaryotic UPS systems. Our findings fill a long-standing gap in cell biology and provide guiding principles for designing more efficient drug delivery vehicles.
153. Targeting Intracellular Protein–Protein Interactions With Macrocyclic Peptides
Marina Buyanova and Dehua Pei, Trends Pharmacol. Sci. 2021, XX, XX, XX-XX, DOI: 10.1016/j.tips.2021.11.008
Intracellular protein–protein interactions (PPIs) are challenging targets for traditional drug modalities. Macrocyclic peptides (MPs) prove highly effective PPI inhibitors in vitro and can be rapidly discovered against PPI targets by rational design or screening combinatorial libraries but are generally impermeable to the cell membrane. Recent advances in MP science and technology are allowing for the development of ‘drug-like’ MPs that potently and specifically modulate intracellular PPI targets in cell culture and animal models. In this review, we highlight recent progress in generating cell-permeable MPs that enter the mammalian cell by passive diffusion, endocytosis followed by endosomal escape, or as-yet unknown mechanisms.
152. Bacterial Toxins Escape the Endosome by Inducing Vesicle Budding and Collapse
Ashweta Sahni and Dehua Pei, ACS Chem. Biol. 2021, 16, 11, 2415–2422, DOI: 10.1021/acschembio.1c00540
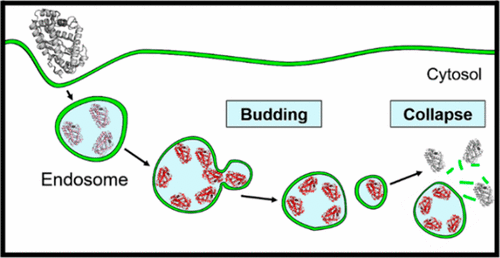
Bacterial protein toxins autonomously enter the cytosol of the target cell where they modify the activities of host components to exert their toxic effects. Many of the toxins enter the host cell by endocytosis followed by endosomal escape. However, their mechanism of endosomal escape remains unresolved. We show herein that diphtheria toxin (DT) and NleC of enteropathogenic Escherichia coli exit the endosome by inducing budding and collapse of small toxin-enriched vesicles from the endosomal membrane.
151. Discovery of a Bicyclic Peptidyl Pan-Ras Inhibitor
Marina Buyanova, Shurui Cai, Jahan Cooper, Curran Rhodes, Heba Salim, Ashweta Sahni, Punit Upadhyaya, Rui Yang, Amar Sarkar, Na Li, Qi-En Wang, and Dehua Pei, J. Med. Chem. 2021, 64, 13038−13053, DOI: 10.1021/acs.jmedchem.1c01130
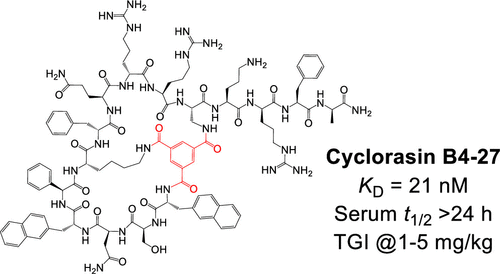
The Ras subfamily of small GTPases is mutated in ∼30% of human cancers and represents compelling yet challenging anticancer drug targets owing to their flat protein surface. We previously reported a bicyclic peptidyl inhibitor, cyclorasin B3, which binds selectively to Ras–GTP with modest affinity and blocks its interaction with downstream effector proteins in vitro but lacks cell permeability or biological activity. In this study, optimization of B3 yielded a potent pan-Ras inhibitor, cyclorasin B4-27, which binds selectively to the GTP-bound forms of wild-type and mutant Ras isoforms (KD = 21 nM for KRasG12V–GppNHp) and is highly cell-permeable and metabolically stable (serum t1/2 > 24 h). B4-27 inhibits Ras signaling in vitro and in vivo by blocking Ras from interacting with downstream effector proteins and induces apoptosis of Ras-mutant cancer cells. When administered systemically (i.v.), B4-27 suppressed tumor growth in two different mouse xenograft models at 1–5 mg/kg of daily doses.
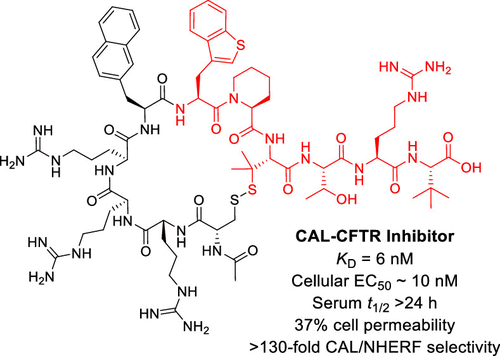
150. Cyclic Peptidyl Inhibitors against CAL/CFTR Interaction for Treatment of Cystic Fibrosis
Patrick G. Dougherty, Jack H. Wellmerling, Amritendu Koley, Jessica K. Lukowski, Amanda B. Hummon, Estelle Cormet-Boyaka, and Dehua Pei, J. Med. Chem., 2020, 63, 15773-15784, DOI: 10.1021/acs.jmedchem.0c01528
Cystic fibrosis (CF) is caused by mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene, encoding for a chloride ion channel. Membrane expression of CFTR is negatively regulated by CFTR-associated ligand (CAL). We previously showed that inhibition of the CFTR/CAL interaction with a cell-permeable peptide improves the function of rescued F508del-CFTR. In this study, optimization of the peptidyl inhibitor yielded PGD97, which exhibits a KD value of 6 nM for the CAL PDZ domain, ≥ 130-fold selectivity over closely related PDZ domains, and a serum t1/2 of >24 h. In patient-derived F508del homozygous cells, PGD97 (100 nM) increased short-circuit currents by ∼3-fold and further potentiated the therapeutic effects of small-molecule correctors (e.g., VX-661) by ∼2-fold (with an EC50 of ∼10 nM). Our results suggest that PGD97 may be used as a novel treatment for CF, either as a single agent or in combination with small-molecule correctors/potentiators.
149. A Peptidyl Inhibitor that Blocks Calcineurin–NFAT Interaction and Prevents Acute Lung Injury
Patrick G. Dougherty, Manjula Karpurapu, Amritendu Koley, Jessica K. Lukowski, Ziqing Qian, Teja Srinivas Nirujogi, Luiza Rusu, Sangwoon Chung, Amanda B. Hummon, Hao W. Li, John W. Christman, and Dehua Pei, J. Med. Chem., 2020, 63, 12853-12872, DOI: 10.1021/acs.jmedchem.0c01236
Acute respiratory distress syndrome (ARDS) is an inflammatory lung disease with a high morbidity and mortality rate, for which no pharmacologic treatment is currently available. Our previous studies discovered that a pivotal step in the disease process is the activation of the nuclear factor of activated T cells (NFAT) c3 in lung macrophages, suggesting that inhibitors against the upstream protein phosphatase calcineurin should be effective for prevention/treatment of ARDS. Herein, we report the development of a highly potent, cell-permeable, and metabolically stable peptidyl inhibitor, CNI103, which selectively blocks the interaction between calcineurin and NFATc3, through computational and medicinal chemistry. CNI103 specifically inhibited calcineurin signaling in vitro and in vivo and exhibited a favorable pharmacokinetic profile, broad tissue distribution following different routes of administration, and minimal toxicity. Our data indicate that CNI103 is a promising novel treatment for ARDS and other inflammatory diseases.

148. Rational design of cell-permeable cyclic peptides containing a D-Pro-L-Pro motif
Jin Wen; Hui Liao; Kye Stachowski; Jordan P. Hempfling; Ziqing Qian; Chunhua Yuan; Mark P. Foster; Dehua Pei, Bioorganic & Medicinal Chemistry, 2020, 28, 1157112, DOI: 10.1016/j.bmc.2020.115711
Cyclic peptides are capable of binding to challenging targets (e.g., proteins involved in protein-protein interactions) with high affinity and specificity, but generally cannot gain access to intracellular targets because of poor membrane permeability. In this work, we discovered a conformationally constrained cyclic cell-penetrating peptide (CPP) containing a D-Pro-L-Pro motif, cyclo(AFΦrpPRRFQ) (where Φ is L-naphthylalanine, r is D-arginine, and p is D-proline). The structural constraints provided by cyclization and the D-Pro-L-Pro motif permitted the rational design of cell-permeable cyclic peptides of large ring sizes (up to 16 amino acids). This strategy was applied to design a potent, cell-permeable, and biologically active cyclic peptidyl inhibitor, cyclo(YpVNFΦrpPRR) (where Yp is L-phosphotyrosine), against the Grb2 SH2 domain. Multidimensional NMR spectroscopic and circular dichroism analyses revealed that the cyclic CPP as well as the Grb2 SH2 inhibitor assume a predominantly random coil structure but have significant β-hairpin character surrounding the D-Pro-L-Pro motif. These results demonstrate cyclo(AFΦrpPRRFQ) as an effective CPP for endocyclic (insertion of cargo into the CPP ring) or exocyclic delivery of biological cargos (attachment of cargo to the Gln side chain).

147. Cell-Penetrating Peptides Escape the Endosome by Inducing Vesicle Budding and Collapse
Ashweta Sahni; Ziqing Qian; Dehua Pei, ACS Chem. Biol. 2020, 15, 2485−2492, DOI: 10.1021/acschembio.0c00478
Cell-penetrating peptides (CPPs) are capable of delivering membrane-impermeable cargoes (including small molecules, peptides, proteins, nucleic acids, and nanoparticles) into the cytosol of mammalian cells and have the potential to revolu-tionize biomedical research and drug discovery. However, the mechanism of action of CPPs has remained poorly under-stood, especially how they escape from the endosome into the cytosol following endocytic uptake. We show herein that CPPs exit the endosome by inducing budding and collapse of CPP-enriched vesicles from the endosomal membrane. This mechanism provides a theoretical basis for designing CPPs and other delivery vehicles of improved efficiencies.


146. Engineering Cell-Permeable Proteins through Insertion of Cell-Penetrating Motifs into Surface Loops
Kuangyu Chen; Dehua Pei, ACS Chem. Biol. 2020, 15, 2568−2576, DOI: 10.1021/acschembio.0c00593
Effective delivery of proteins into the cytosol of mammalian cells would open the door to a wide range of applications. However, despite great efforts from numerous investigators, effective protein delivery in a clinical setting is yet to be accomplished. Herein we report a potentially general approach to engineering cell-permeable proteins by genetically grafting a short cell-penetrating peptide (CPP) to an exposed loop of a protein of interest. The grafted peptide is conformationally constrained, exhibiting enhanced proteolytic stability and cellular entry efficiency. Applying this technique to enhanced green fluorescent protein (EGFP), protein-tyrosine phosphatase 1B (PTP1B), and purine nucleoside phosphorylase (PNP) rendered all three proteins cell-permeable and biologically active in cellular assays. When added into growth medium at 0.5-5 μM concentrations, the engineered PTP1B dose-dependently reduced the phosphotyrosine levels of intracellular proteins, while the modified PNP corrected the metabolic deficiency of PNP-deficient mouse T lymphocytes, providing a potential enzyme replacement therapy for a rare genetic disease.

145. Enhancing the Cell-Permeability of Stapled Peptides with a Cyclic Cell-Penetrating Peptide
Patrick G Dougherty; Jin Wen; Xiaoyan Pan; Amritendu Koley; Jian-Guo Ren; Ashweta Sahni; Ruchira Basu; Heba Salim; George Appiah-Kubi; Ziqing Qian; Dehua Pei, J. Med. Chem. 2019, 62, 10098−10107, DOI: 10.1021/acs.jmedchem.9b00456
Stapled peptides recapitulate the binding affinity and specificity of α-helices in proteins, resist proteolytic degradation, and may provide a novel modality against challenging drug targets such as protein-protein interactions. However, most of the stapled peptides have limited cell-permeability or are impermeable to the cell membrane. We show herein that stapled peptides can be rendered highly cell-permeable by conjugating a cyclic cell-penetrating peptide to their N-terminus, C-terminus, or stapling unit. Application of this strategy to two previously reported, membrane-impermeable peptidyl inhibitors against the MDM2/p53 and β-catenin/TCF interactions resulted in the generation of potent proof-of-concept anti-proliferative agents against key therapeutic targets.

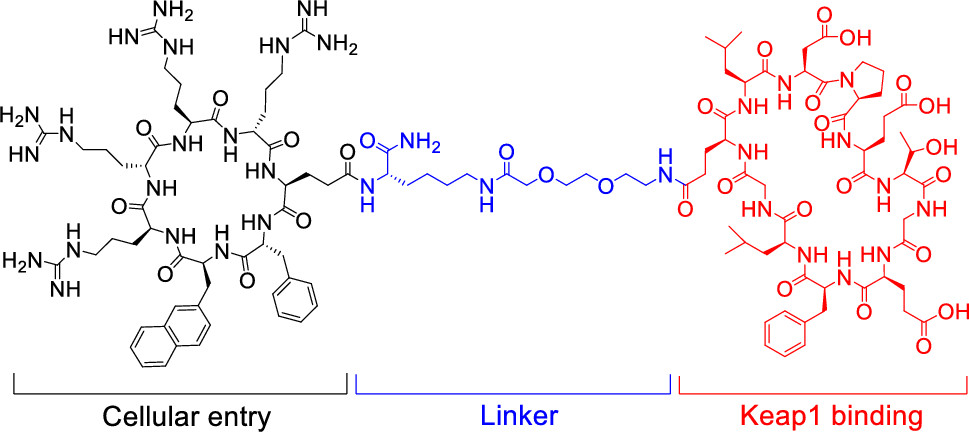
144. Development of a Cell-Permeable Cyclic Peptidyl Inhibitor against the Keap1–Nrf2 Interaction
Heba Salim; Jian Song; Ashweta Sahni; Dehua Pei, J. Org. Chem. 2020, 85, 1416−1424, DOI: 10.1021/acs.joc.9b02367
Macrocyclic peptides have proven to be highly effective inhibitors of protein–protein interactions but generally lack cell permeability to access intracellular targets. We show herein that macrocyclic peptides may be rendered highly cell-permeable and biologically active by conjugating them with a cyclic cell-penetrating peptide (CPP). A previously reported cyclic peptidyl inhibitor against the Kelch-like ECH-associated protein 1 (Keap1)–nuclear factor erythroid-2 (Nrf2) interaction (KD = 18 nM) was covalently attached to a cyclic CPP through a flexible linker. The resulting bicyclic peptide retained the Keap1-binding activity, resisted proteolytic degradation, readily entered mammalian cells, and activated the transcriptional activity of Nrf2 at nanomolar to low micromolar concentrations in cell culture. The inhibitor provides a useful tool for investigating the biological function of Keap1–Nrf2 and a potential lead for further development into a novel class of anti-inflammatory and anticancer agents. Our data suggest that other membrane-impermeable cyclic peptides may be similarly rendered cell-permeable by conjugation with a cyclic CPP.
143. Cyclic Cell-Penetrating Peptides with Single Hydrophobic Groups
Dehua Pei; Jian Song; Ziqing Qian; Ashweta Sahni; Kuangyu Chen; ChemBioChem 2019, 20,2085 –2088, DOI: 10.1002/cbic.201900370
A new family of cyclic cell‐penetrating peptides (CPPs) is discovered and differs from previously reported cyclic CPPs by containing only a single hydrophobic residue. The optimal CPP structure consists of four arginine residues and a hydrophobic residue of long alkyl chain (e.g., a decyl group) in a cyclohexapeptide ring. The most active member of this family, CPP 17, has an intrinsic cellular entry efficiency similar to that of cyclic CPP12, the most active CPP reported to date. However, CPP 17 is 2.8‐fold more active than CPP12 under high serum protein concentrations, presumably because of less protein binding. CPP 17 enters the cell primarily by direct translocation at relatively low concentration (≥5 μM).
142. Understanding Cell Penetration of Cyclic Peptides
Patrick G. Dougherty; Ashweta Sahni; Dehua Pei; Chem. Rev. 2019, 119, 17, 10241–10287, DOI: 10.1021/acs.chemrev.9b00008
Approximately 75% of all disease-relevant human proteins, including those involved in intracellular protein–protein interactions (PPIs), are undruggable with the current drug modalities (i.e., small molecules and biologics). Macrocyclic peptides provide a potential solution to these undruggable targets because their larger sizes (relative to conventional small molecules) endow them the capability of binding to flat PPI interfaces with antibody-like affinity and specificity. Powerful combinatorial library technologies have been developed to routinely identify cyclic peptides as potent, specific inhibitors against proteins including PPI targets. However, with the exception of a very small set of sequences, the vast majority of cyclic peptides are impermeable to the cell membrane, preventing their application against intracellular targets. This Review examines common structural features that render most cyclic peptides membrane impermeable, as well as the unique features that allow the minority of sequences to enter the cell interior by passive diffusion, endocytosis/endosomal escape, or other mechanisms. We also present the current state of knowledge about the molecular mechanisms of cell penetration, the various strategies for designing cell-permeable, biologically active cyclic peptides against intracellular targets, and the assay methods available to quantify their cell-permeability.
141. Crystal structure of the Redβ C-terminal domain in complex with λ Exonuclease reveals an unexpected homology with λ Orf and an interaction with Escherichia coli single stranded DNA binding protein
Bacteriophage λ encodes a DNA recombination system that includes a 5′-3′ exonuclease (λ Exo) and a single strand annealing protein (Redβ). The two proteins form a complex that is thought to mediate loading of Redβ directly onto the single-stranded 3′-overhang generated by λ Exo. Here, we present a 2.3 Å crystal structure of the λ Exo trimer bound to three copies of the Redβ C-terminal domain (CTD). Mutation of residues at the hydrophobic core of the interface disrupts complex formation in vitro and impairs recombination in vivo. The Redβ CTD forms a three-helix bundle with unexpected structural homology to phage λ Orf, a protein that binds to E. coli single-stranded DNA binding protein (SSB) to function as a recombination mediator. Based on this relationship, we found that Redβ binds to full-length SSB, and to a peptide corresponding to its nine C-terminal residues, in an interaction that requires the CTD. These results suggest a dual role of the CTD, first in binding to λ Exo to facilitate loading of Redβ directly onto the initial single-stranded DNA (ssDNA) at a 3′-overhang, and second in binding to SSB to facilitate annealing of the overhang to SSB-coated ssDNA at the replication fork.
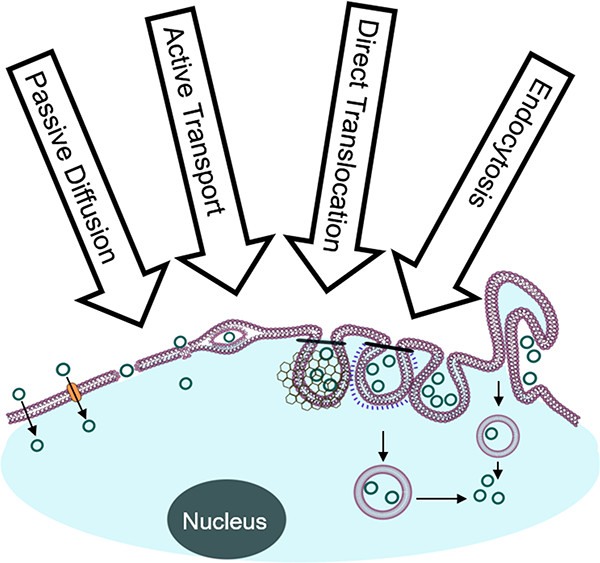
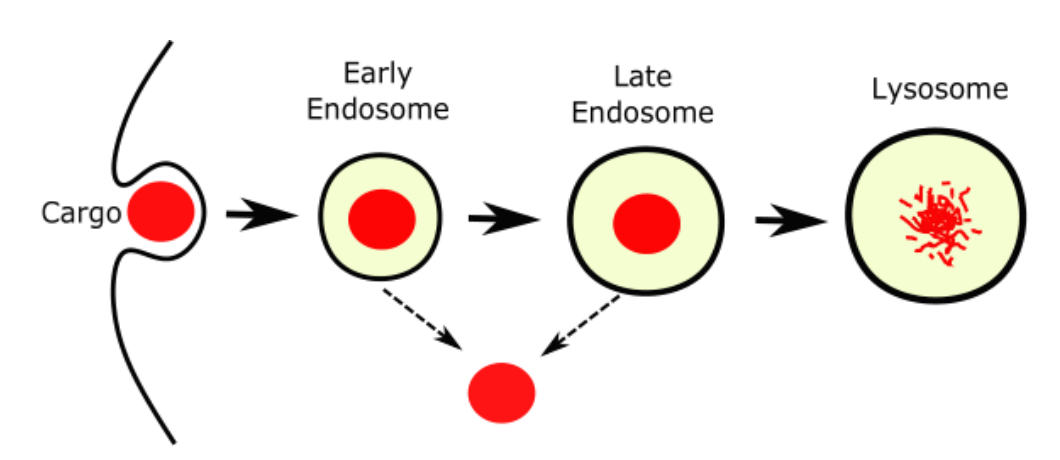
140. Overcoming Endosomal Entrapment in Drug Delivery
Dehua Pei and Marina Buyanova; Bioconjugate Chemistry; 2019, 302273-283; DOI: 10.1021/acs.bioconjchem.8b00778
Intracellular delivery of biological agents such as peptides, proteins, and nucleic acids generally rely on the endocytic pathway as the major uptake mechanism, resulting in their entrapment inside
the endosome and lysosome. The recent discovery of cell-penetrating molecules of exceptionally high endosomal escape and cytosolic delivery efficiencies and elucidation of their mechanism of
action represent major breakthroughs in this field. In this Topical Review, we provide an overview of the recent progress in understanding and enhancing the endosomal escape process and the new opportunities opened up by these recent findings.
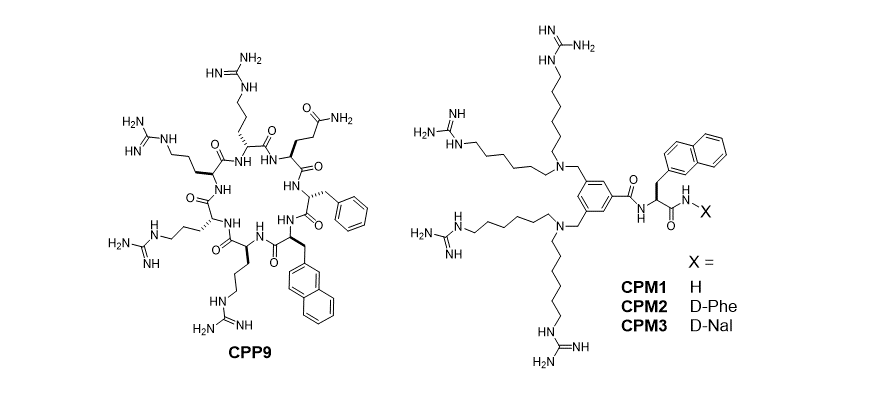
139. Non‐Peptidic Cell‐Penetrating Motifs for Mitochondrion‐Specific Cargo Delivery
George Appiah Kubi, Dr. Ziqing Qian, Dr. Souad Amiar, Ashweta Sahni, Prof. Dr. Robert V. Stahelin, Prof. Dr. Dehua Pei; Angewandte Chemie. 10.1002/ange.201811940.
Mitochondrial dysfunction is linked to a variety of human illnesses, but selective delivery of therapeutics into the mitochondrion is challenging. Now a family of amphipathic cell‐penetrating motifs (CPMs) is presented, consisting of four guanidinium groups and one or two aromatic hydrophobic groups (naphthalene) assembled through a central scaffold (a benzene ring). The CPMs and CPM‐cargo conjugates efficiently enter the interior of cultured mammalian cells and are specifically localized into the mitochondrial matrix, as revealed by high‐resolution confocal microscopy. With a membrane‐impermeable peptide as cargo, the CPMs exhibited ≥170‐fold higher delivery efficiency than previous mitochondrial delivery vehicles. Conjugation of a small‐molecule inhibitor of heat shock protein 90 to a CPM resulted in accumulation of the inhibitor inside the mitochondrial matrix with greatly enhanced anticancer activity. The CPMs showed minimal effect on the viability or the mitochondrial membrane potential of mammalian cells.
138. Cell-Permeable Bicyclic Peptidyl Inhibitors against NEMO-IκB Kinase Interaction Directly from a Combinatorial Library
Curran A. Rhodes, Patrick G. Dougherty, Jahan K. Cooper, Ziqing Qian, Steffen Lindert, Qi-En Wang, and Dehua Pei; J. Am. Chem. Soc., 2018, 140 (38), pp 12102–12110; DOI: 10.1021/jacs.8b06738
Macrocyclic peptides are capable of binding to flat protein surfaces such as the interfaces of protein–protein interactions with antibody-like affinity and specificity, but generally lack cell permeability in order to access intracellular targets. In this work, we designed and synthesized a large combinatorial library of cell-permeable bicyclic peptides, in which the first ring consisted of randomized peptide sequences for potential binding to a target of interest, while the second ring featured a family of different cell-penetrating motifs, for both cell penetration and target binding. The library was screened against the IκB kinase α/β (IKKα/β)-binding domain of NF-κB essential modulator (NEMO), resulting in the discovery of several cell-permeable bicyclic peptides, which inhibited the NEMO-IKKβ interaction with low μM IC50 values. Further optimization of one of the hits led to a relatively potent and cell-permeable NEMO inhibitor (IC50 = 1.0 μM), which selectively inhibited canonical NF-κB signaling in mammalian cells and the proliferation of cisplatin-resistant ovarian cancer cells. The inhibitor provides a useful tool for investigating the biological functions of NEMO/NF-κB and a potential lead for further development of a novel class of anti-inflammatory and anticancer drugs.

137. Inhibition of nuclear factor of activated T cells (NFAT) c3 activation attenuates acute lung injury and pulmonary edema in murine models of sepsis
Manjula Karpurapu, Yong Gyu Lee, Ziqing Qian, Jin Wen, Megan N. Ballinger, Luiza Rusu, Sangwoon Chung, Jing Deng, Feng Qian, Brenda F. Reader, Teja Srinivas Nirujogi, Gye Young Park, Dehua Pei and John W. Christman; Oncotarget. 2018; 9:10606-10620; DOI: 10.18632/oncotarget.24320
Specific therapies targeting cellular and molecular events of sepsis induced Acute Lung Injury (ALI) pathogenesis are lacking. We have reported a pivotal role for Nuclear Factors of Activated T cells (NFATc3) in regulating macrophage phenotype during sepsis induced ALI and subsequent studies demonstrate that NFATc3 transcriptionally regulates macrophage CCR2 and TNFα gene expression. Mouse pulmonary microvascular endothelial cell monolayer maintained a tighter barrier function when co-cultured with LPS stimulated NFATc3 deficient macrophages whereas wild type macrophages caused leaky monolayer barrier. More importantly, NFATc3 deficient mice showed decreased neutrophilic lung inflammation, improved alveolar capillary barrier function, arterial oxygen saturation and survival benefit in lethal CLP sepsis mouse models. In addition, survival of wild type mice subjected to the lethal CLP sepsis was not improved with broad-spectrum antibiotics, whereas the survival of NFATc3 deficient mice was improved to 40–60% when treated with imipenem. Passive adoptive transfer of NFATc3 deficient macrophages conferred protection against LPS induced ALI in wild type mice. Furthermore, CP9-ZIZIT, a highly potent, cell-permeable peptide inhibitor of Calcineurin inhibited NFATc3 activation. CP9-ZIZIT effectively reduced sepsis induced inflammatory cytokines and pulmonary edema in mice. Thus, this study demonstrates that inhibition of NFATc3 activation by CP9-ZIZIT provides a potential therapeutic option for attenuating sepsis induced ALI/pulmonary edema.
136. Cell-permeable bicyclic peptidyl inhibitors against T-cell protein tyrosine phosphatase from a combinatorial library
Hui Liao and Dehua Pei; Org. Biomol. Chem., 2017, 15, 9595-9598
Protein tyrosine phosphatases (PTPs) have been challenging targets for inhibitor design, because all PTPs share a highly conserved active site structure, which is positively charged and requires negatively charged moieties for tight binding. In this study, we developed cell-permeable bicyclic peptidyl inhibitors against T-cell PTP (TCPTP), which feature a cell-penetrating motif in one ring and a target-binding sequence in the second ring.
135. Bicyclic Peptides as Next‐Generation Therapeutics
Curran A. Rhodes and Dehua Pei; Chem. Eur. J. 2017, 23, 12690 – 12703
Bicyclic peptides have greater conformational rigidity and metabolic stability than linear and monocyclic peptides and are capable of binding to challenging drug targets with antibody‐like affinity and specificity. Powerful combinatorial library technologies have recently been developed to rapidly synthesize and screen large bicyclic peptide libraries for ligands against enzymes, receptors, and protein–protein interaction targets. Bicyclic peptides have been developed as potential therapeutics against a wide range of diseases, drug targeting agents, imaging/diagnostic probes, and research tools. In this Minireview, we provide a summary of the recent progresses on the synthesis and applications of bicyclic peptides.
134. Generation of a cell-permeable cycloheptapeptidyl inhibitor against the peptidyl–prolyl isomerase Pin1
Walaa Bedewy, Hui Liao, Nageh A. Abou-Taleb, Sherif F. Hammad, Tamer Nasr and Dehua Pei Org. Biomol. Chem. 2017 DOI: 10.1039/c7ob00430c
Cyclic peptides are capable of binding and modulating challenging drug targets including protein–protein interactions. However, their lack of membrane permeability prevents their application against intracellular targets. In this study, we show that it is possible to design a cell-permeable and biologically active cycloheptapeptide inhibitor against the intracellular enzyme peptidyl–prolyl isomerase Pin1 by integrating cell-penetrating and target-binding sequences.
133. Enhancing the Cell Permeability and Metabolic Stability of Peptidyl Drugs by Reversible Bicyclization
Ziqing Qian, Curran A. Rhodes, Lucas C. McCroskey, Jin Wen, George Appiah-Kubi, David J. Wang, Denis C. Guttridge and Dehua Pei Angew. Chem. Int. Ed. 2016 DOI: 10.1002/anie.201610888
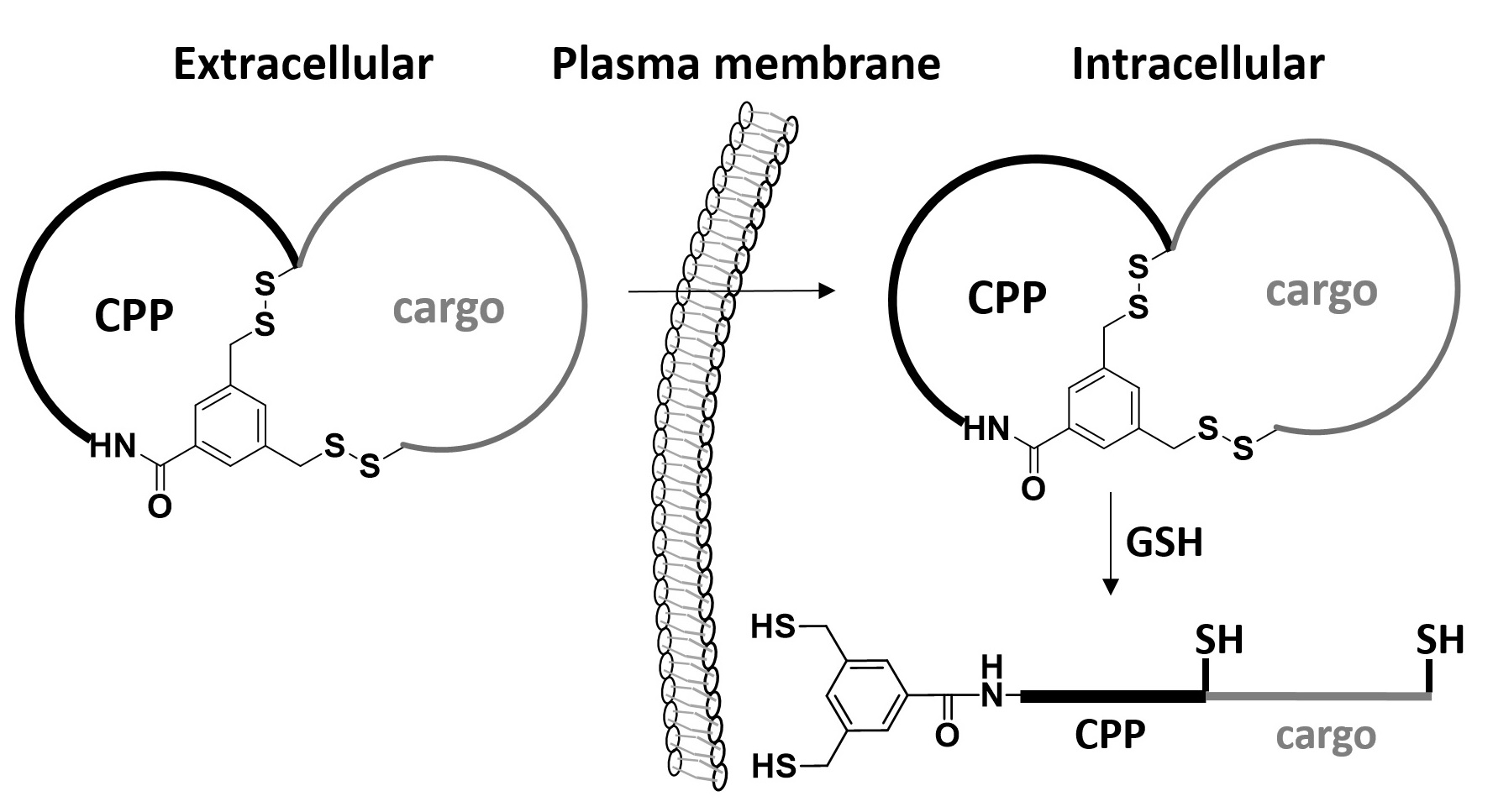
Therapeutic applications of peptides are currently limited by their proteolytic instability and impermeability to the cell membrane. A general, reversible bicyclization strategy is now reported to increase both the proteolytic stability and cell permeability of peptidyl drugs. A peptide drug is fused with a short cell-penetrating motif and converted into a conformationally constrained bicyclic structure through the formation of a pair of disulfide bonds. The resulting bicyclic peptide has greatly enhanced proteolytic stability as well as cell-permeability. Once inside the cell, the disulfide bonds are reduced to produce a linear, biologically active peptide. This strategy was applied to generate a cell-permeable bicyclic peptidyl inhibitor against the NEMO-IKK interaction.
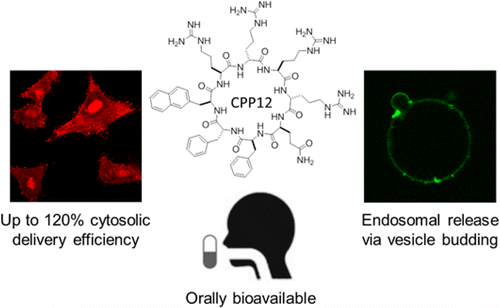
132. Discovery and Mechanism of Highly Efficient Cyclic Cell-Penetrating Peptides
Ziqing Qian, Agnieszka Martyna, Ryan L. Hard, Jiang Wang, George Appiah-Kubi, Christopher Coss, Mitch A. Phelps, Jeremy S. Rossman, and Dehua Pei Biochemistry 2016 DOI: 10.1021/acs.biochem.6b00226.
Previous cell-penetrating peptides (CPPs) generally have low cytosolic delivery efficiencies, because of inefficient endosomal escape. In this study, a family of small, amphipathic cyclic peptides was found to be highly efficient CPPs, with cytosolic delivery efficiencies of up to 120% (compared to 2.0% for Tat). These cyclic CPPs bind directly to the plasma membrane phospholipids and enter mammalian cells via endocytosis, followed by efficient release from the endosome. Their total cellular uptake efficiency correlates positively with the binding affinity for the plasma membrane, whereas their endosomal escape efficiency increases with the endosomal membrane-binding affinity. The cyclic CPPs induce membrane curvature on giant unilamellar vesicles and budding of small vesicles, which subsequently collapse into amorphous lipid/peptide aggregates. These data suggest that cyclic CPPs exit the endosome by binding to the endosomal membrane and inducing CPP-enriched lipid domains to bud off as small vesicles. Together with their high proteolytic stability, low cytotoxicity, and oral bioavailability, these cyclic CPPs should provide a powerful system for intracellular delivery of therapeutic agents and chemical probes.
131. LNK/SH2B3 regulates IL-7 receptor signaling in normal and malignant B-progenitors
Ying Cheng, Kudakwashe Chikwava, Chao Wu, Haibing Zhang, Anchit Bhagat, Dehua Pei, John K. Choi, and Wei Tong J. Clin. Invest. 2016 DOI: 10.1172/JCI81468.
Philadelphia chromosome–like acute lymphoblastic leukemia (Ph-like ALL) is a high-risk ALL commonly associated with alterations that affect the tyrosine kinase pathway, tumor suppressors, and lymphoid transcription factors. Loss-of-function mutations in the gene-encoding adaptor protein LNK (also known as SH2B3) are found in Ph-like ALLs; however, it is not clear how LNK regulates normal B cell development or promotes leukemogenesis. Here, we have shown that combined loss of Lnk and tumor suppressors Tp53 or Ink4a/Arf in mice triggers a highly aggressive and transplantable precursor B-ALL. Tp53–/–Lnk–/– B-ALLs displayed similar gene expression profiles to human Ph-like B-ALLs, supporting use of this model for preclinical and molecular studies. Preleukemic Tp53–/–Lnk–/– pro-B progenitors were hypersensitive to IL-7, exhibited marked self-renewal in vitro and in vivo, and were able to initiate B-ALL in transplant recipients. Mechanistically, we demonstrated that LNK regulates pro-B progenitor homeostasis by attenuating IL-7–stimuated JAK/STAT5 signaling via a direct interaction with phosphorylated JAK3. Moreover, JAK inhibitors were effective in prolonging survival of mice transplanted with Lnk–/–Tp53–/– leukemia. Additionally, synergistic administration of PI3K/mTOR and JAK inhibitors further abrogated leukemia development. Hence, our results suggest that LNK suppresses IL-7R/JAK/STAT signaling to restrict pro-/pre-B progenitor expansion and leukemia development, providing a pathogenic mechanism and a potential therapeutic approach for B-ALLs with LNK mutations.
130. Discovery of a Direct Ras Inhibitor by Screening a Combinatorial Library of Cell-Permeable Bicyclic Peptides
Thi B. Trinh, Punit Upadhyaya, Ziqing Qian, and Dehua Pei. ACS Comb. Sci. 2015 DOI: 10.1021/acscombsci.5b00164
Cyclic peptides have great potential as therapeutic agents and research tools. However, their applications against intracellular targets have been limited, because cyclic peptides are generally impermeable to the cell membrane. It was previously shown that fusion of cyclic peptides with a cyclic cell-penetrating peptide resulted in cell-permeable bicyclic peptides that are proteolytically stable and biologically active in cellular assays. In this work, we tested the generality of the bicyclic approach by synthesizing a combinatorial library of 5.7 x 106 bicyclic peptides featuring a degenerate sequence in the first ring and an invariant cell-penetrating peptide in the second ring. Screening of the library against oncoprotein K-Ras G12V followed by hit optimization produced a moderately potent and cell-permeable K-Ras inhibitor, which physically blocks the Ras-effector interactions in vitro, inhibits the signaling events downstream of Ras in cancer cells, and induces apoptosis of the cancer cells. Our approach should be generally applicable to developing cell-permeable bicyclic peptide inhibitors against other intracellular proteins.
129. Screening One-Bead-One-Compound Peptide Libraries for Optimal Kinase Substrates
Thi B. Trinh and Dehua Pei. Methods in Molecular Biology 2015 1360: 169-181
128. Direct Inhibitors of Ras-Effector Protein Interactions
Punit Upadhyaya, Walaa Bedewy, and Dehua Pei. Mini-Rev. Med. Chem., 2015,PMID:26423701 .
Activating Ras mutations are associated with ~30% of all human cancers, which often respond poorly to standard therapies. The four Ras isoforms are therefore highly attractive targets for anticancer drug discovery. However, Rasproteins function through protein–protein interactions and their surfaces lack any major pockets for small molecules to bind; as a result they have been declared “undruggable” for the past 30 years. Several breakthroughs during the past few years may finally remove Ras from the list of undruggable proteins. This mini-review discusses the current approaches to developing inhibitors especially cyclic peptides that physically block the interaction between Ras and its downstream effector proteins, which is potentially the most effective approach for treating Ras mutant cancers.
127. A Selective, Cell-Permeable Nonphosphorylated Bicyclic Peptidyl Inhibitor against Peptidyl–Prolyl Isomerase Pin1
Bisheng Jiang and Dehua Pei. J. Med. Chem., 2015, 58, 6306-6312.
Pin1 regulates the levels and functions of phosphoproteins by catalyzing phosphorylation-dependent cis/trans isomerization of peptidyl–prolyl bonds. Previous Pin1 inhibitors contained phosphoamino acids, which are metabolically unstable and have poor membrane permeability. In this work, we report a cell-permeable and metabolically stable nonphosphorylated bicyclic peptide as a potent and selective Pin1 inhibitor, which inhibited the intracellular Pin1 activity in cultured mammalian cells but had little effect on other isomerases such as Pin4, FKBP12, or cyclophilin A.
126. Inhibition of Ras Signaling by Blocking Ras–Effector Interactions with Cyclic Peptides
Punit Upadhyaya, Ziqing Qian, Nicholas G. Selner, Sarah R. Clippinger, Zhengrong Wu, Roger Briesewitz, and Dehua Pei. Angew. Chem. Int. Ed., 2015, 54, 7602-7606.
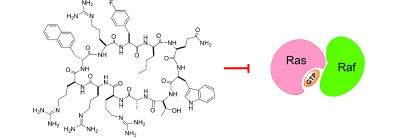
Ras genes are frequently activated in human cancers, but the mutant Ras proteins remain largely “undruggable” through the conventional small-molecule approach owing to the absence of any obvious binding pockets on their surfaces. By screening a combinatorial peptide library, followed by structure–activity relationship (SAR) analysis, we discovered a family of cyclic peptides possessing both Ras-binding and cell-penetrating properties. These cell-permeable cyclic peptides inhibit Ras signaling by binding to Ras-GTP and blocking its interaction with downstream proteins and they induce apoptosis of cancer cells. Our results demonstrate the feasibility of developing cyclic peptides for the inhibition of intracellular protein–protein interactions and of direct Ras inhibitors as a novel class of anticancer agents.
125. Intracellular Delivery of Peptidyl Ligands by Reversible Cyclization: Discovery of a PDZ Domain Inhibitor that Rescues CFTR Activity
Ziqing Qian, Xiaohua Xu, Jeanine F. Amacher, Dean R. Madden, Estelle Cormet-Boyaka, and Dehua Pei. Angew. Chem. Int. Ed., 2015, 54, 5874-5878.
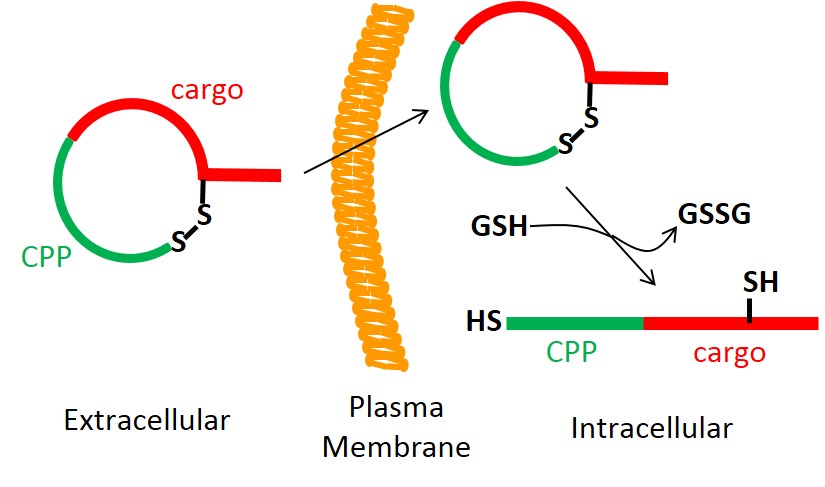
A general strategy was developed for the intracellular delivery of linear peptidyl ligands through fusion to a cell-penetrating peptide and cyclization of the fusion peptides via a disulfide bond. The resulting cyclic peptides are cell permeable and have improved proteolytic stability. Once inside the cell, the disulfide bond is reduced to produce linear biologically active peptides. This strategy was applied to generate a cell-permeable peptide substrate for real-time detection of intracellular caspase activities during apoptosis and an inhibitor for the CFTR-associated ligand (CAL) PDZ domain as a potential treatment for cystic fibrosis.
124. Monitoring the cytosolic entry of cell-penetrating peptides using a pH-sensitive fluorophore
Ziqing Qian, Patrick G. Dougherty, and Dehua Pei. Chem. Commun., 2015, 51, 2162-2165
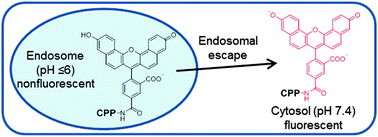
We report a simple, effective method to assess the cytosolic delivery efficiency and kinetics of cell-penetrating peptides using a pH-sensitive fluorescent probe, naphthofluorescein.
123. Synthesis and Screening of One-Bead-One-Compound Cyclic Peptide Libraries
Ziqing Qian, Punit Upadhyaya, and Dehua Pei. Methods in Molecular Biology 2015 1248: 39-53
122. Direct Ras Inhibitors Identified from a Structurally Rigidified Bicyclic Peptide Library
Punit Upadhyaya, Ziqing Qian, Nurlaila A.A. Habir, and Dehua Pei. Tetrahedron 2014, 70(42), 7714-7720
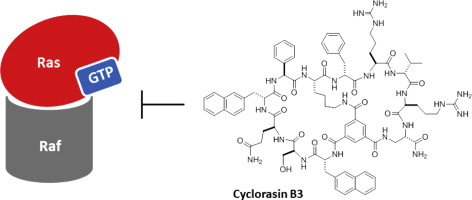
A one-bead-two-compound (OBTC) library of structurally rigidified bicyclic peptides was chemically synthesized on TentaGel microbeads (90 μm), with each bead displaying a unique bicyclic peptide on its surface and a linear encoding peptide of the same sequence in its interior. Screening of the library against oncogenic K-Ras G12V mutant identified two classes of Ras ligands. The class I ligands apparently bind to the effector-binding site and inhibit the Ras–Raf interaction, whereas the class II ligand appears to bind to a yet unidentified site different from the effector-binding site. These Ras ligands provide useful research tools and may be further developed into therapeutic agents.
121. Structure-Based Optimization of a Peptidyl Inhibitor against Calcineurin-Nuclear Factor of Activated T Cell (NFAT) Interaction
Ziqing Qian, Patrick G. Dougherty, Tao Liu, Shameema Oottikkal, Patrick G. Hogan, Christopher M. Hadad, and Dehua Pei. J. Med. Chem., 2014, 57 (18), 7792–7797
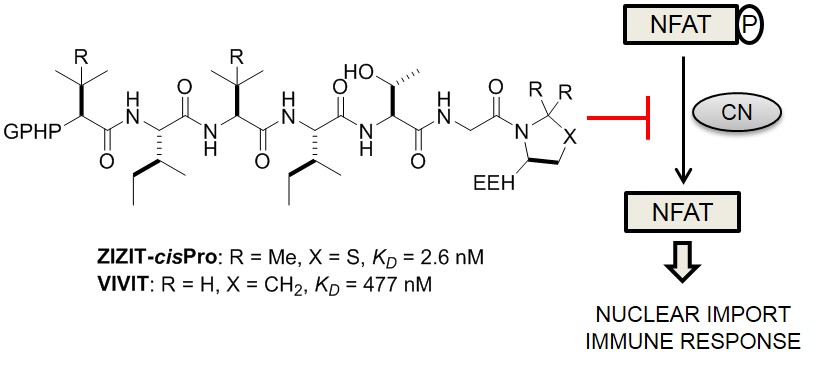
Calcineurin inhibitors such as cyclosporine A and FK506 are effective immunosuppressants but produce severe side effects. Rational modification of a previously reported peptide inhibitor, GPHPVIVITGPHEE (KD ∼ 500 nM), by replacing the two valine residues with tert-leucine and the C-terminal proline with a cis-proline analogue, gave an improved inhibitor ZIZIT-cisPro, which binds to calcineurin with a KD value of 2.6 nM and is more resistant to proteolysis.
120. Cell-Permeable Bicyclic Peptide Inhibitors against Intracellular Proteins
Wenlong Lian, Bisheng Jiang, Ziqing Qian, and Dehua Pei. J. Am. Chem. Soc., 2014, 136 (28), 9830–9833
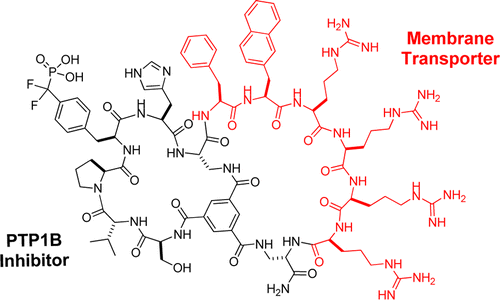
Cyclic peptides have great potential as therapeutic agents and research tools but are generally impermeable to the cell membrane. Fusion of cyclic peptides with a cyclic cell-penetrating peptide produces bicyclic peptides that are cell-permeable and retain the ability to recognize specific intracellular targets. Application of this strategy to protein tyrosine phosphatase 1B and a peptidyl-prolyl cis−trans isomerase (Pin1) isomerase resulted in potent, selective, proteolytically stable, and biologically active inhibitors against the enzymes.
119. Early Endosomal Escape of a Cyclic Cell-Penetrating Peptide Allows Effective Cytosolic Cargo Delivery
Ziqing Qian, Jonathan R. LaRochelle, Bisheng Jiang, Wenlong Lian, Ryan L. Hard, Nicholas G. Selner, Rinrada Luechapanichkul, Amy M. Barrios, and Dehua Pei. Biochemistry, 2014, 53 (24), 4034–4046
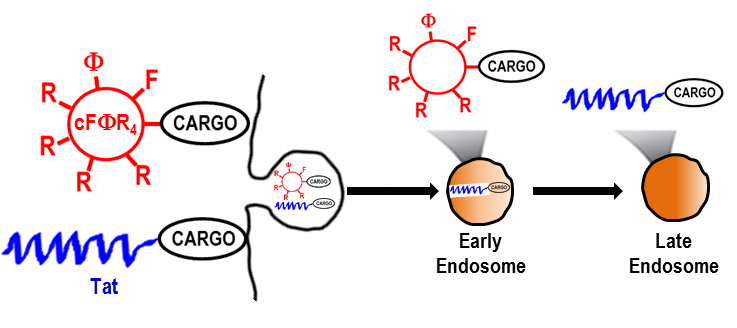
Cyclic heptapeptide cyclo(FΦRRRRQ) (cFΦR4, where Φ is l-2-naphthylalanine) was recently found to be efficiently internalized by mammalian cells. In this study, its mechanism of internalization was investigated by perturbing various endocytic events through the introduction of pharmacologic agents and genetic mutations. The results show that cFΦR4 binds directly to membrane phospholipids, is internalized into human cancer cells through endocytosis, and escapes from early endosomes into the cytoplasm. Its cargo capacity was examined with a wide variety of molecules, including small-molecule dyes, linear and cyclic peptides of various charged states, and proteins. Depending on the nature of the cargos, they may be delivered by endocyclic (insertion of cargo into the cFΦR4 ring), exocyclic (attachment of cargo to the Gln side chain), or bicyclic approaches (fusion of cFΦR4 and cyclic cargo rings). The overall delivery efficiency (i.e., delivery of cargo into the cytoplasm and nucleus) of cFΦR4 was 4–12-fold higher than those of nonaarginine, HIV Tat-derived peptide, or penetratin. The higher delivery efficiency, coupled with superior serum stability, minimal toxicity, and synthetic accessibility, renders cFΦR4 a useful transporter for intracellular cargo delivery and a suitable system for investigating the mechanism of endosomal escape.
118. Diverse Levels of Sequence Selectivity and Catalytic Efficiency of Protein-Tyrosine Phosphatases
Nicholas G. Selner, Rinrada Luechapanichkul, Xianwen Chen, Benjamin G. Neel, Zhong-Yin Zhang, Stefan Knapp, Charles E. Bell, and Dehua Pei. Biochemistry, 2014, 53 (2), 397–412
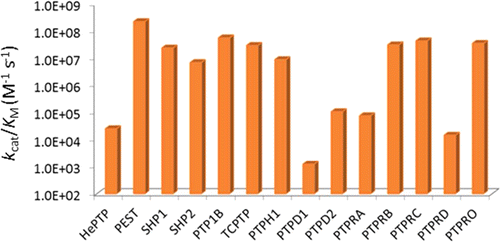
The sequence selectivity of 14 classical protein-tyrosine phosphatases (PTPs) (PTPRA, PTPRB, PTPRC, PTPRD, PTPRO, PTP1B, SHP-1, SHP-2, HePTP, PTP-PEST, TCPTP, PTPH1, PTPD1, and PTPD2) was systematically profiled by screening their catalytic domains against combinatorial peptide libraries. All of the PTPs exhibit similar preference for pY peptides rich in acidic amino acids and disfavor positively charged sequences but differ vastly in their degrees of preference/disfavor. Some PTPs (PTP-PEST, SHP-1, and SHP-2) are highly selective for acidic over basic (or neutral) peptides (by >105-fold), whereas others (PTPRA and PTPRD) show no to little sequence selectivity. PTPs also have diverse intrinsic catalytic efficiencies (kcat/KM values against optimal substrates), which differ by >105-fold due to different kcat and/or KM values. Moreover, PTPs show little positional preference for the acidic residues relative to the pY residue. Mutation of Arg47 of PTP1B, which is located near the pY-1 and pY-2 residues of a bound substrate, decreased the enzymatic activity by 3–18-fold toward all pY substrates containing acidic residues anywhere within the pY-6 to pY+5 region. Similarly, mutation of Arg24, which is situated near the C-terminus of a bound substrate, adversely affected the kinetic activity of all acidic substrates. A cocrystal structure of PTP1B bound with a nephrin pY1193 peptide suggests that Arg24 engages in electrostatic interactions with acidic residues at the pY+1, pY+2, and likely other positions. These results suggest that long-range electrostatic interactions between positively charged residues near the PTP active site and acidic residues on pY substrates allow a PTP to bind acidic substrates with similar affinities, and the varying levels of preference for acidic sequences by different PTPs are likely caused by the different electrostatic potentials near their active sites. The implications of the varying sequence selectivity and intrinsic catalytic activities with respect to PTP in vivo substrate specificity and biological functions are discussed.
117. Screening Bicyclic Peptide Libraries for Protein–Protein Interaction Inhibitors: Discovery of a Tumor Necrosis Factor-α Antagonist
Wenlong Lian, Punit Upadhyaya, Curran A. Rhodes, Yusen Liu, and Dehua Pei. J. Am. Chem. Soc., 2013, 135 (32), 11990–11995
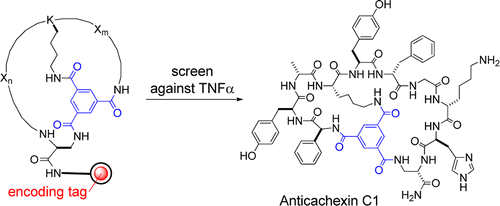
Protein–protein interactions represent a new class of exciting but challenging drug targets, because their large, flat binding sites lack well-defined pockets for small molecules to bind. We report here a methodology for chemical synthesis and screening of large combinatorial libraries of bicyclic peptides displayed on rigid small-molecule scaffolds. With planar trimesic acid as the scaffold, the resulting bicyclic peptides are effective for binding to protein surfaces such as the interfaces of protein–protein interactions. Screening of a bicyclic peptide library against tumor necrosis factor-α (TNFα) identified a potent antagonist that inhibits the TNFα–TNFα receptor interaction and protects cells from TNFα-induced cell death. Bicyclic peptides of this type may provide a general solution for inhibition of protein–protein interactions.
116. Profiling the Substrate Specificity of Protein Kinases by On-Bead Screening of Peptide Libraries
Thi B. Trinh, Qing Xiao, and Dehua Pei. Biochemistry, 2013, 52 (33), 5645–5655
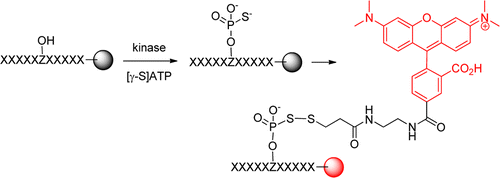
A robust, high-throughput method has been developed to screen one-bead–one-compound peptide libraries to systematically profile the sequence specificity of protein kinases. Its ability to provide individual sequences of the preferred substrates permits the identification of sequence contextual effects and nonpermissive residues. Application of the library method to kinases Pim1, MKK6, and Csk revealed that Pim1 and Csk are highly active toward peptide substrates and recognize specific sequence motifs, whereas MKK6 has little activity or sequence selectivity against peptide substrates. Pim1 recognizes peptide substrates of the consensus RXR(H/R)X(S/T); it accepts essentially any amino acid at the S/T–2 and S/T+1 positions, but strongly disfavors acidic residues (Asp or Glu) at the S/T–2 position and a proline residue at the S/T+1 position. The selected Csk substrates show strong sequence covariance and fall into two classes with the consensus sequences of (D/E)EPIYϕXϕ and (D/E)(E/D)S(E/D/I)YϕXϕ (where X is any amino acid and ϕ is a hydrophobic amino acid). Database searches and in vitro kinase assays identified phosphatase PTP-PEST as a Pim1 substrate and phosphatase SHP-1 as a potential Csk substrate. Our results demonstrate that the sequence specificity of protein kinases is defined not only by favorable interactions between permissive residue(s) on the substrate and their cognate binding site(s) on the kinase but also by repulsive interactions between the kinase and nonpermissive residues
115. Specificity Profiling of Protein Phosphatases toward Phosphoseryl and Phosphothreonyl Peptides
Qing Xiao, Rinrada Luechapanichkul, Yujing Zhai, and Dehua Pei. J. Am. Chem. Soc., 2013, 135 (26), 9760–9767
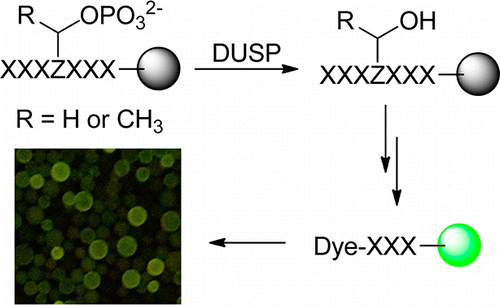
A combinatorial library method was developed to systematically profile the substrate specificity of protein phosphatases toward phosphoseryl (pS) and phosphothreonyl (pT) peptides. Application of this method and a previously reported phosphotyrosyl (pY) library screening technique to dual-specificity phosphatase (DUSP) VH1 of vaccinia virus revealed that VH1 is highly active toward both pS/pT and pY peptides. VH1 exhibits different and more stringent sequence specificity toward pS/pT than pY substrates. Unlike previously characterized protein tyrosine phosphatases (PTPs), the activity and specificity of VH1 are primarily determined by the amino acid residues C-terminal to the pS, pT, or pY residue. In contrast, the mammalian VH1-related (VHR) DUSP has intrinsically low catalytic activity toward pS and pT substrates, suggesting that its primary physiological function is to dephosphorylate pY residues in substrate proteins. This method is applicable to other DUSPs and protein-serine/threonine phosphatases, and the substrate specificity data will be useful for identifying the physiological substrates of these enzymes.
114. Inhibition of Ras–effector Interactions by Cyclic Peptides
Xianghong Wu, Punit Upadhyaya, Miguel A. Villalona-Calero, Roger Briesewitz, and Dehua Pei. Med. Chem. Comm., 2013, 4, 378-382
A combinatorial library of 6 × 106 cyclic peptides was synthesized in the one bead-two compound format, with each bead displaying a unique cyclic peptide on its surface and a linear peptide encoding tag in its interior. Screening of the library against K-Ras identified compounds that bound K-Ras with submicromolar affinity and disrupted its interaction with effector proteins.
113. Specificity Profiling of Dual Specificity Phosphatase Vaccinia VH1-related (VHR) Reveals Two Distinct Substrate Binding Modes
Rinrada Luechapanichkul, Xianwen Chen, Hashem A. Taha, Shubham Vyas, Xiaoyan Guan, Michael A. Freitas, Chrstopher M. Hadad, and Dehua Pei. J. Biol. Chem. 2013, 288(9), 6498-6510.
Vaccinia VH1-related (VHR) is a dual specificity phosphatase that consists of only a single catalytic domain. Although several protein substrates have been identified for VHR, the elements that control the in vivo substrate specificity of this enzyme remain unclear. In this work, the in vitro substrate specificity of VHR was systematically profiled by screening combinatorial peptide libraries. VHR exhibits more stringent substrate specificity than classical protein-tyrosine phosphatases and recognizes two distinct classes of Tyr(P) peptides. The class I substrates are similar to the Tyr(P) motifs derived from the VHR protein substrates, having sequences of (D/E/φ)(D/S/N/T/E)(P/I/M/S/A/V)pY(G/A/S/Q) or (D/E/φ)(T/S)(D/E)pY(G/A/S/Q) (where φ is a hydrophobic amino acid and pY is phosphotyrosine). The class II substrates have the consensus sequence of (V/A)P(I/L/M/V/F)X1–6pY (where X is any amino acid) with V/A preferably at the N terminus of the peptide. Site-directed mutagenesis and molecular modeling studies suggest that the class II peptides bind to VHR in an opposite orientation relative to the canonical binding mode of the class I substrates. In this alternative binding mode, the Tyr(P) side chain binds to the active site pocket, but the N terminus of the peptide interacts with the carboxylate side chain of Asp164, which normally interacts with the Tyr(P) + 3 residue of a class I substrate. Proteins containing the class II motifs are efficient VHR substrates in vitro, suggesting that VHR may act on a novel class of yet unidentified Tyr(P) proteins in vivo.
112. Systematic Characterization of the Specificity of the SH2 Domains of Cytoplasmic Tyrosine Kinases
Bing Zhao, Pauline H. Tan, Shawn S.C. Li, and Dehua Pei. J Proteomics. 2013, 81, 56-69.
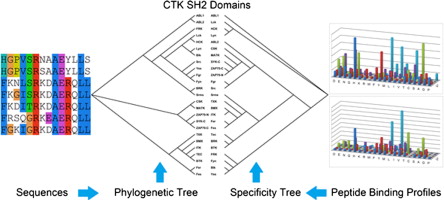
Cytoplasmic tyrosine kinases (CTK) generally contain a Src-homology 2 (SH2) domain, whose role in the CTK family is not fully understood. Here we report the determination of the specificity of 25 CTK SH2 domains by screening one-bead-one-compound (OBOC) peptide libraries. Based on the peptide sequences selected by the SH2 domains, we built Support Vector Machine (SVM) models for the prediction of binding ligands for the SH2 domains. These models yielded support for the progressive phosphorylation model for CTKs in which the overlapping specificity of the CTK SH2 and kinase domains has been proposed to facilitate targeting of the CTK substrates with at least two potential phosphotyrosine (pTyr) sites. We curated 93 CTK substrates with at least two pTyr sites catalyzed by the same CTK, and showed that 71% of these substrates had at least two pTyr sites predicted to bind a common CTK SH2 domain. More importantly, we found 34 instances where there was at least one pTyr site predicted to be recognized by the SH2 domain of the same CTK, suggesting that the SH2 and kinase domains of the CTKs may cooperate to achieve progressive phosphorylation of a protein substrate.
111. Global Analysis of Peptide Cyclization Efficiency
Amit Thakkar, Thi B. Trinh, and Dehua Pei. ACS Comb. Sci., 2013, 15 (2), 120–129
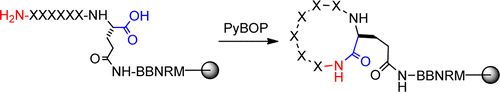
Cyclic peptides are of considerable interest in drug discovery and nanotechnology. However, macrocyclization of peptides and other compounds has often been perceived as synthetically challenging and the cyclization yields are affected by several factors including the ring size, peptide sequence, and the reaction conditions. Through the screening of combinatorial peptide libraries, we analyzed the cyclization efficiency of >2 million peptide sequences to determine the effect of ring size, peptide sequence, and solvent on the backbone (N-to-C) cyclization of peptides. Our results show that on-resin cyclization of medium- and large-sized rings (cyclohexapeptides and above) with PyBOP is essentially quantitative for ≥99.96% of the sequences, with small amounts of dimer formation observed for <4% of these sequences. Cyclization of small rings (cyclotetrapeptides and cyclopentapeptides) is considerably more difficult and accompanied by significant cyclic dimer formation. Peptides that are difficult to cyclize are generally rich in Lys(Boc) and Arg(Pbf) residues as well as sterically hindered residues [e.g., Thr(tBu)] at the N-terminus. The majority of these difficult sequences can be cyclized to completion by the addition of aqueous additives to the cyclization reaction.
110. Efficient Delivery of Cyclic Peptides into Mammalian Cells with Short Sequence Motifs
Ziqing Qian, Tao Liu, Yu-Yu Liu, Roger Briesewitz, Amy M. Barrios, Sissy M. Jhiang, and Dehua Pei. ACS Chem. Biol., 2013, 8 (2), 423–431
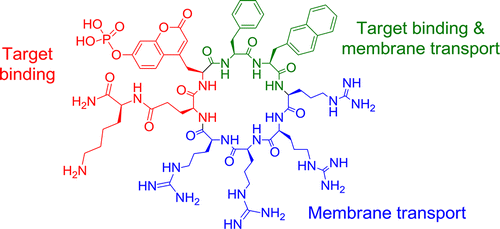
Cyclic peptides hold great potential as therapeutic agents and research tools, but their broad application has been limited by poor membrane permeability. Here, we report a potentially general approach for intracellular delivery of cyclic peptides. Short peptide motifs rich in arginine and hydrophobic residues (e.g., FΦRRRR, where Φ is l-2-naphthylalanine), when embedded into small- to medium-sized cyclic peptides (7–13 amino acids), bound to the plasma membrane of mammalian cultured cells and were subsequently internalized by the cells. Confocal microscopy and a newly developed peptide internalization assay demonstrated that cyclic peptides containing these transporter motifs were translocated into the cytoplasm and nucleus at efficiencies 2–5-fold higher than that of nonaarginine (R9). Furthermore, incorporation of the FΦRRRR motif into a cyclic peptide containing a phosphocoumaryl aminopropionic acid (pCAP) residue generated a cell permeable, fluorogenic probe for detecting intracellular protein tyrosine phosphatase activities.